Download as docx, pdf, or txt
You might also like
- Epigenetics How Environment Changes Your BiologyDocument144 pagesEpigenetics How Environment Changes Your BiologyAdje100% (1)
- Embryology Inderbir Singh PDFDocument370 pagesEmbryology Inderbir Singh PDFbiswas soma56% (9)
- Enzymes Review Worksheet: Name: . DateDocument4 pagesEnzymes Review Worksheet: Name: . Dateapi-233187566100% (1)
- Pathophysiology of Osteomyelitis DiagramDocument1 pagePathophysiology of Osteomyelitis DiagramKim Enrico JumarangNo ratings yet
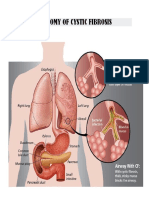
- Anatomy of Cystic FibrosisDocument5 pagesAnatomy of Cystic FibrosislazylawatudentNo ratings yet
- Pathophysiology Schistosomiasis: Table in New WindowDocument7 pagesPathophysiology Schistosomiasis: Table in New WindowKaren Leigh MagsinoNo ratings yet
- Phosphate Imbalances PDFDocument1 pagePhosphate Imbalances PDFKaye RicoNo ratings yet
- PATHOPHYSIOLOGYDocument4 pagesPATHOPHYSIOLOGYJasonlee BaluyotNo ratings yet
- Ateneo de Davao University College of NursingDocument161 pagesAteneo de Davao University College of Nursingnevajane100% (1)
- Bea-Case StudyDocument21 pagesBea-Case Studybea pegadNo ratings yet
- Cardiovascular Agents ReviewerDocument18 pagesCardiovascular Agents ReviewerJoycel CeñidozaNo ratings yet
- Acute Gastroenteritis With Some DehydrationDocument6 pagesAcute Gastroenteritis With Some DehydrationKiyla920% (1)
- Transcultural Nursing Quiz 2Document2 pagesTranscultural Nursing Quiz 2Jenny AjocNo ratings yet
- Nursing Theory PDFDocument4 pagesNursing Theory PDFBSN 1-YA-22 ELITIONG, MARY JEAN A.No ratings yet
- Acute Upper Airway ObstructionDocument2 pagesAcute Upper Airway ObstructionBudy CaecarianNo ratings yet
- NCM 111 Nursing Research Reviewer PrelimsDocument8 pagesNCM 111 Nursing Research Reviewer PrelimsKIRSTEN CHAVEZNo ratings yet
- 4 Nursing Research Alpha 2017Document8 pages4 Nursing Research Alpha 2017Nicole DimarucutNo ratings yet
- Notes Stress Adaptation PDFDocument19 pagesNotes Stress Adaptation PDFAnil PatelNo ratings yet
- MCN Lec Fetal DevpDocument580 pagesMCN Lec Fetal DevpknotstmNo ratings yet
- Community Acquired Pneumonia Concept MapDocument1 pageCommunity Acquired Pneumonia Concept MapSebastianNo ratings yet
- Pernicious AnemiaDocument7 pagesPernicious AnemiaTracy PearlNo ratings yet
- Concept Map COPDDocument2 pagesConcept Map COPDMatthew Frank Melendez QuerolNo ratings yet
- Pathophysiology of Upper Gastrointestinal BleedingDocument1 pagePathophysiology of Upper Gastrointestinal BleedingkimmybapkiddingNo ratings yet
- That Will Affect The Choice of Teaching Strategy or ApproachDocument2 pagesThat Will Affect The Choice of Teaching Strategy or ApproachDagmawit GirmaNo ratings yet
- 8. Care of Child With GI Dysfunction (1) ءءءءDocument44 pages8. Care of Child With GI Dysfunction (1) ءءءءNuhaNo ratings yet
- NCM 104 Cellular Aberration Lecture 2007Document104 pagesNCM 104 Cellular Aberration Lecture 2007Kris TejereroNo ratings yet
- Castro, Almeacor D. - Digestive ExerciseDocument4 pagesCastro, Almeacor D. - Digestive ExerciseJaja Dofitas CastroNo ratings yet
- Course Description: 3. Communities (Community Service)Document35 pagesCourse Description: 3. Communities (Community Service)April Mae Magos LabradorNo ratings yet
- Cholelithiasis Brief DiscussionDocument8 pagesCholelithiasis Brief Discussionriel100% (6)
- 2010 TFN Module Unit 06 Nursing Paradigm - NursingDocument43 pages2010 TFN Module Unit 06 Nursing Paradigm - NursingCamille Denise Nucum100% (1)
- Philippine Nursing Law of 2002 Ra # 9173Document15 pagesPhilippine Nursing Law of 2002 Ra # 9173Michael Maunda AmpuanNo ratings yet
- Vitamin KDocument6 pagesVitamin KCaesarioNo ratings yet
- 6-Health Problems Common in PreschoolerDocument36 pages6-Health Problems Common in PreschoolerPam Lala100% (2)
- Pathophysiology of DiarrheaDocument3 pagesPathophysiology of DiarrheaFathur RahmatNo ratings yet
- BLOOD DYSCRASIA ADocument2 pagesBLOOD DYSCRASIA AMyami Bersamen100% (1)
- Diarrhea: Practice Essentials, Background, PathophysiologyDocument6 pagesDiarrhea: Practice Essentials, Background, PathophysiologyBilal El BariNo ratings yet
- FCM 1.6 - NFEP (Filariasis)Document10 pagesFCM 1.6 - NFEP (Filariasis)ZazaNo ratings yet
- Application of Research in Nursing Leadership and ManagementDocument25 pagesApplication of Research in Nursing Leadership and ManagementLee BuelaNo ratings yet
- Drug StudyDocument6 pagesDrug StudyLizli LoredoNo ratings yet
- El Filibusterismo (Summary)Document4 pagesEl Filibusterismo (Summary)Nestor Bong Bordaje NemeñoNo ratings yet
- AppendicitisDocument11 pagesAppendicitisbobtagubaNo ratings yet
- MCN Lect Module 2.5 Complications of Labor and Birth 1 and 2Document7 pagesMCN Lect Module 2.5 Complications of Labor and Birth 1 and 2AmethystNo ratings yet
- Antepartum Intrapartum: Preparing For The BabyDocument3 pagesAntepartum Intrapartum: Preparing For The BabyMikhaela Andree MarianoNo ratings yet
- Pathophysiology Acute Bacterial MeningitisDocument2 pagesPathophysiology Acute Bacterial MeningitisNadira Farah PrayogoNo ratings yet
- Individual PediaDocument37 pagesIndividual PediaLianna M. MilitanteNo ratings yet
- Drug Study Medcor AguinaldoDocument6 pagesDrug Study Medcor AguinaldoYana PotNo ratings yet
- Ulcerative ColitisDocument9 pagesUlcerative Colitiskint manlangitNo ratings yet
- Typhoid FeverDocument1 pageTyphoid FeverMarkChesterSaguidNagen100% (1)
- 7 PathophysiologyDocument1 page7 PathophysiologyKizzel MacapazNo ratings yet
- CiprobayDocument2 pagesCiprobayianecunar100% (1)
- Post-Partum Hemorrhage Pathophysiology PaperDocument5 pagesPost-Partum Hemorrhage Pathophysiology Paperapi-399619969No ratings yet
- Care of Clients With Problems in OxygenationDocument282 pagesCare of Clients With Problems in OxygenationAYTONA, JAMAICA F.No ratings yet
- A Case Presentation On AgiosarcomaDocument48 pagesA Case Presentation On AgiosarcomaAsniah Hadjiadatu AbdullahNo ratings yet
- Pathophy of Appendicitis (Or) by Mizzy BaylonDocument2 pagesPathophy of Appendicitis (Or) by Mizzy BaylonmizzybaylonNo ratings yet
- Reflection PaperDocument2 pagesReflection PapershanoiapowelllNo ratings yet
- Community Acquired Pneumonia, A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsFrom EverandCommunity Acquired Pneumonia, A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsNo ratings yet
- The politics of hunger: Protest, poverty and policy in England, <i>c.</i> 1750–<i>c.</i> 1840From EverandThe politics of hunger: Protest, poverty and policy in England, <i>c.</i> 1750–<i>c.</i> 1840No ratings yet
- Ward Class On Cystic FibrosisDocument58 pagesWard Class On Cystic FibrosisJannen CasasNo ratings yet
- Cystic Fibrosis NotesDocument1 pageCystic Fibrosis Notesmanaal2728No ratings yet
- Cystic FibrosisDocument1 pageCystic FibrosisRubz JeanNo ratings yet
- Cysticfibrosis 150401114224 Conversion Gate01Document29 pagesCysticfibrosis 150401114224 Conversion Gate01Marwan SweityNo ratings yet
- Chapter 259. Cystic FibrosisDocument7 pagesChapter 259. Cystic FibrosisAntónio CarvalhoNo ratings yet
- Cystic FibrosisDocument20 pagesCystic FibrosisIndrajit Barua100% (1)
- Cystic FibrosisDocument20 pagesCystic FibrosisIndrajit BaruaNo ratings yet
- Thyroidectomy NCPDocument3 pagesThyroidectomy NCPKim Enrico Jumarang100% (2)
- Aging ProcessDocument17 pagesAging ProcessKim Enrico JumarangNo ratings yet
- OsteomyelitisDocument13 pagesOsteomyelitisKim Enrico JumarangNo ratings yet
- Hypertension: Jumarang, Kim Enrico M. BSN401 STI - Global CityDocument6 pagesHypertension: Jumarang, Kim Enrico M. BSN401 STI - Global CityKim Enrico JumarangNo ratings yet
- Oesteomyelitis: By: Llana, Arnold Jay Jumarang, Kim Enrico MDocument5 pagesOesteomyelitis: By: Llana, Arnold Jay Jumarang, Kim Enrico MKim Enrico JumarangNo ratings yet
- RH Disease (Erythroblastosis Fetalis) : Jumarang, Kim Enrico M. Micropara Bsn301Document4 pagesRH Disease (Erythroblastosis Fetalis) : Jumarang, Kim Enrico M. Micropara Bsn301Kim Enrico JumarangNo ratings yet
- Diabetes: Jumarang, Kim Enrico M. BSN401 STI - Global CityDocument5 pagesDiabetes: Jumarang, Kim Enrico M. BSN401 STI - Global CityKim Enrico JumarangNo ratings yet
- Acute Lymphocytic LeukemiaDocument7 pagesAcute Lymphocytic LeukemiaKim Enrico JumarangNo ratings yet
- Cell Structure and FunctionDocument66 pagesCell Structure and FunctionMark Anthony CorpuzNo ratings yet
- Protease Opt Opt1Document23 pagesProtease Opt Opt1Youness ElbabouriNo ratings yet
- Barr Body: Chromatin Is An Inactive X Chromosome in A Cell With More ThanDocument4 pagesBarr Body: Chromatin Is An Inactive X Chromosome in A Cell With More Thanhasan jamiNo ratings yet
- N - and C-Terminal Protein Sequencing by MALDI ISD Feb 2009Document5 pagesN - and C-Terminal Protein Sequencing by MALDI ISD Feb 2009andrewmunk100% (2)
- Chapter 2Document62 pagesChapter 2GG MMNo ratings yet
- Biology Solomon Full ChapterDocument67 pagesBiology Solomon Full Chaptermilton.alvarado146100% (5)
- Activity 28 - Adaptive ImmunityDocument6 pagesActivity 28 - Adaptive ImmunityKaren Joy MagbanuaNo ratings yet
- Measuring Water Potential in Potato Tubers LABDocument10 pagesMeasuring Water Potential in Potato Tubers LABMonica Paris SisourathNo ratings yet
- Microbial Physiology. Microbial Metabolism. Enzymes. Nutrition. Bioenergetics. Bacterial Growth and MultiplicationDocument102 pagesMicrobial Physiology. Microbial Metabolism. Enzymes. Nutrition. Bioenergetics. Bacterial Growth and MultiplicationamitNo ratings yet
- Mitosis Vs Meiosis ReadingDocument3 pagesMitosis Vs Meiosis Readingshazia imamNo ratings yet
- Extra Questions, CELL, CLASS-9Document17 pagesExtra Questions, CELL, CLASS-9Raman Jena0% (1)
- Principles of Genetics Lecture 20 Transduction - Mapping The OriginDocument4 pagesPrinciples of Genetics Lecture 20 Transduction - Mapping The OriginmeilunlyNo ratings yet
- Epq JDocument16 pagesEpq JHenry Paul OjedaNo ratings yet
- Action Potentials and Synapses HandoutsDocument6 pagesAction Potentials and Synapses HandoutsKelly TrainorNo ratings yet
- Chemistry and Fundamental BiotechnologyDocument3 pagesChemistry and Fundamental BiotechnologyRaweeha SaifNo ratings yet
- Anemia in SepsisDocument7 pagesAnemia in SepsismedusineNo ratings yet
- Role of Sigma FactorDocument29 pagesRole of Sigma Factorsandhya namadaraNo ratings yet
- 11Document15 pages11AmaniNo ratings yet
- AlBorg ResultSubServicesDocument1 pageAlBorg ResultSubServicesxwxwwx57No ratings yet
- Enzymes NotesDocument46 pagesEnzymes NotesCuthbert MweembaNo ratings yet
- ShohayokDocument36 pagesShohayokSafiqa TasfiaNo ratings yet
- Inside The Cell: Chapter 5: The Last Chapter: Cell Aging and DeathDocument13 pagesInside The Cell: Chapter 5: The Last Chapter: Cell Aging and DeathAnonymous LXV7PwpNo ratings yet
- These GNYUBKIN Vasily 2015Document151 pagesThese GNYUBKIN Vasily 2015AliNo ratings yet
- ACROBiosystems Product CatalogueDocument40 pagesACROBiosystems Product CatalogueArunee HansawongsakulNo ratings yet
- Set 4 Cell Biology Grades 4 6Document13 pagesSet 4 Cell Biology Grades 4 6Diva NoorzaiNo ratings yet
- Lipids LehningerDocument7 pagesLipids LehningerElla BangalanNo ratings yet
- 2.2 Pentose and Uronic PathwayDocument28 pages2.2 Pentose and Uronic PathwayYared gebremedhinNo ratings yet