
Professional Documents
Culture Documents
Wade Organic Chemistry A II Bottom 415 1091
Wade Organic Chemistry A II Bottom 415 1091
Uploaded by
溫緯中Copyright:
Available Formats
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5822)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Mpemba Effect When Can Hot Water Freeze Faster Than ColdDocument10 pagesThe Mpemba Effect When Can Hot Water Freeze Faster Than Cold陳琮方No ratings yet
- Assignment 10Document3 pagesAssignment 10Ayush shuklaNo ratings yet
- Total HardnessDocument6 pagesTotal HardnesspermatakomputerNo ratings yet
- CS095 Membrane ElectrolyzerDocument13 pagesCS095 Membrane ElectrolyzerJuan Diego Arbelaez AlzateNo ratings yet
- 04 (2) HJBHDocument3 pages04 (2) HJBHncubepharmaNo ratings yet
- Explanation SnowDocument2 pagesExplanation SnowAhmad RamadanNo ratings yet
- Mse HWDocument2 pagesMse HWJessica AguilarNo ratings yet
- Written Exam Supramolecular Chemistry Winter 2018 F. Diederich, Y. YamakoshiDocument5 pagesWritten Exam Supramolecular Chemistry Winter 2018 F. Diederich, Y. YamakoshiSelim RezaNo ratings yet
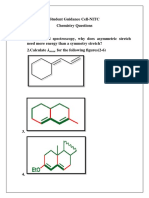
- Student Guidance Cell-NITC Chemistry QuestionsDocument5 pagesStudent Guidance Cell-NITC Chemistry QuestionsVedurupaka Venkata SaiNo ratings yet
- Good Paper!!!Document9 pagesGood Paper!!!muriloinnocentiniNo ratings yet
- Essentials of Heat and Fluid Flow in Porous Media: PrefaceDocument3 pagesEssentials of Heat and Fluid Flow in Porous Media: PrefaceAbdul Rauf AttariNo ratings yet
- Olga Afa PDFDocument91 pagesOlga Afa PDFSofiane BouradaNo ratings yet
- DiffusionDocument2 pagesDiffusionKrishna Kumar MishraNo ratings yet
- C H E M 1 0 1: Chem 101-General Chemistry Energy & ChemistryDocument46 pagesC H E M 1 0 1: Chem 101-General Chemistry Energy & ChemistryRyo SumidaNo ratings yet
- USP-NF Atorvastatin Calcium TabletsDocument9 pagesUSP-NF Atorvastatin Calcium TabletsPhạm Đức LộcNo ratings yet
- Faraday's Law On ElectrolysisDocument2 pagesFaraday's Law On ElectrolysisFavourNo ratings yet
- Methoxyphenol (Although 3-Allyl-4-Methoxyphenol Can't Be Excluded Either) - Thus, The ProtonDocument5 pagesMethoxyphenol (Although 3-Allyl-4-Methoxyphenol Can't Be Excluded Either) - Thus, The ProtonAnonymous f8P42082UNo ratings yet
- Salt Analysis Notes 12Document42 pagesSalt Analysis Notes 12allancholan200609No ratings yet
- Measurement Systems: Application and Design by Ernest O. DoebelinDocument19 pagesMeasurement Systems: Application and Design by Ernest O. DoebelinAlhji AhmedNo ratings yet
- Adnan Aljarallah 1988 Kinetic of MTBE Over AmberlystDocument6 pagesAdnan Aljarallah 1988 Kinetic of MTBE Over AmberlystJason NunezNo ratings yet
- Introduction To Power PlantDocument31 pagesIntroduction To Power PlantLofi Radio100% (1)
- UV-Vis Application - Quantitative Analysis Using Second-Order Derivative Spectrum No A349Document2 pagesUV-Vis Application - Quantitative Analysis Using Second-Order Derivative Spectrum No A349Ramon Trinidad De la ONo ratings yet
- Introduction To ChemistryDocument116 pagesIntroduction To ChemistryJohn Michael SomorostroNo ratings yet
- Water Conveyance With Syphons: September, 2000 (Rev 2009)Document19 pagesWater Conveyance With Syphons: September, 2000 (Rev 2009)Sameer ShrivastavaNo ratings yet
- Silicon Photonics: ApplicationsDocument7 pagesSilicon Photonics: ApplicationscadmoscelticNo ratings yet
- Detailed Lesson Plan in ScienceDocument4 pagesDetailed Lesson Plan in ScienceAdrian TastarNo ratings yet
- Dapus TerbaruDocument4 pagesDapus TerbaruElvina iskandarNo ratings yet
- Astm D6038 - 2014Document4 pagesAstm D6038 - 2014alferedNo ratings yet
- Principles of Food and Bioprocess Engineering (FS 231) Problems On Heat TransferDocument8 pagesPrinciples of Food and Bioprocess Engineering (FS 231) Problems On Heat Transferx비No ratings yet
- Chapter 2-Atomic Structure Worksheet AnswersDocument2 pagesChapter 2-Atomic Structure Worksheet AnswershomamunfatNo ratings yet






































































Wade Organic Chemistry A II Bottom 415 1091
Wade Organic Chemistry A II Bottom 415 1091
Uploaded by
溫緯中Original Description:
Original Title
Copyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
Wade Organic Chemistry A II Bottom 415 1091
Wade Organic Chemistry A II Bottom 415 1091
Uploaded by
溫緯中Copyright:
Available Formats
Dept.Chem.
FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
200
ORGANIC CHEMISTRY- 2
nd
semester
~this handout is only used for education~
Organic Chemistry, Wade, L. G., 8
th
ed., 2013.
Chapter 13 Nuclear Magnetic Resonance Spectroscopy
NMR is the most powerful tool available for organic structure determination. Like, infrared
spectroscopy, NMR can be used with a very small sample, and it done not harm sample. NMR is
used to study a wide variety of nuclei, including
1
H,
13
C,
15
N,
19
F, and
31
P. Organic chemists find
proton (
1
H) and carbon (
13
C) NMR to be most powerful because hydrogen and carbon are major
components of organic compounds.
13.2 Theory of nuclear magnetic resonance
A nucleus with an odd atomic number or an odd mass number has a nuclear spin that can be
observed by NMR spectrometer. A proton is the simplest nucleus, and its odd atomic number of 1
implies it has a spin. The magnetic moment, also called magnetic field, is generated from a
spinning proton as a rotating sphere of positive charge.
Wade, p. 563.
Same as a small bar magnet, when a proton is placed in an external magnetic field (B
0
). The
protons magnetic moment will be aligned either with the external field, or against the field.
Wade, p. 564.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
201
Wade, p. 564.
The lower-energy state with the proton aligned with the field called the alpha-spin (o-spin) state.
The higher-energy state with the proton aligned against the external magnetic field is called the
beta-spin (|-spin).
Wade, p. 564.
In the absence of an external magnetic field, proton magnetic moments have random orientation.
When an external magnetic field is applied, each proton in a sample assumes the o state and |-state.
The energy difference is proportional to the strength of the magnetic field, as expected in the
equation, shown below.
A E =energy difference between o and | spins.
h =Plancks constant
B
0
=strength of the external magnetic field
=gyromagnetic ratio, 26,753 sec
1
gauss
1
for a proton
Magnetic field are measured in guess. The strength if the earths magnetic field is about 0.57 gauss.
The unit tesla (T) is 10,000 gauss.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
202
13.3 Magnetic shielding by electrons
Real protons in organic compounds are not naked. They are surrounded by electrons that partially
shield them from the external magnetic field. The electrons circulate and generate a small induced
magnetic field that opposes the externally applied field.
The result is that the magnetic field at the nucleus is weaker than the external field, and we say the
nucleus is shielded. In methanol, the electronegative oxygen atom withdraws some electron density
from around the hydroxyl proton. The hydroxyl proton is not shielded as much as the methyl protons,
so the hydroxyl proton absorbs at a lower field than the methyl protons. We say that the hydroxyl
proton is deshielded somewhat by the presence of the electronegative oxygen atom.
Wade, 8
th
, p.567. (less shielded =deshielded)
Some facts needed to be understood before we continue
1. The number of different absorptions (also called signal or peaks) implies how many different
types of protons are present.
2. The amount of shielding shown by these absorptions implies the electronic structure of the
molecules close to each type of proton.
3. Theintensities of the signals imply how many protons of each type are present.
4. The splitting of the signals gives information about other nearby protons.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
203
13.4 The NMR spectrometer
Four parts are included in a NMR spectrometer
1. A magnet, with sensitive controller to produce a precise magnetic field
2. A radio-frewuency (RF) transmitter, emitting a precise frequency.
3. A detector, to measure the samples absorption RF energy
4. A recorder, to plot the output from the detector versus
Higher values of the magnetic field are toward the right (upfield), and lower values are toward the
left (downfield). The absorption of more shielded protons appear upfield, toward the right of the
spectrum, and more deshielded protons appear downfield.
13.5 The Chemical Shift
The most common NMR reference compound is tetramethylsilane (CH
3
)
4
Si, abbreviated TMS.
Because silicon is less electronegative than carbon, the methyl groups of TMS are relatively
electron-rich, and their protons are well shielded, compared to most of the organic compounds. Most
of the organic compounds are hydrocarbon or hydrocarbon attached with heteroatoms, such as O, S,
N.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
204
Chemical shift (ppm) =shift downfield from TMS (Hz) / total spectrometer frequency (MHz)
The most common scale of chemical shifts is the o (delta) scale, which we use. The signal from TMS
is defined as 0.00 ppm on the o scale. Most protons of organic compounds are more deshielded than
TMS, so the o scale increase toward the left of the spectrum.
For example; 2130 Hz in a 300 MHz NMR, the ppm is
2130 Hz/300 MHz =7.10 ppm.
However, what frequency will be if it is measured in 600 MHz? Ans: 4260 Hz.
What will influence the values of chemical shift?
As we mentioned the organic compounds sometimes attached some heteroatoms. You may come up
with the idea that is electronegativity.
As you could see, the closer to the bromide atom, the more downfield is the chemical shift (or the
more deshielded is the proton).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
205
Typical values of chemical shifts are what you need to remember!!!
Vinyl and aromatic protons
Double bonds and aromatic rings produce large deshielding effects on their vinyl and aromatic
protons. Benzene absorbs at o 7.2. The methyl protons are deshielded by a small amount, absorbing
at o 2.3.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
206
With this deshi8elding effect, most vinyl protons absorb in the range of o 5 ~6.
Wade, 8
th
, p. 573
Acetylenic hydrogens
Acetylenic hydrogens absorb around o 2.5, compared with o 5 ~6 for vinyl protons.
Aldehyde protons (-CHO) absor at even lower fields than vinyl protons and aromatic protons:
between o 9 ~10. The aldehyde proton is deshielded both by the circulation of the electrons in the
double bond and by the inductive electron-withdrawing effect of the carbonyl oxygen atom.
Hydrogen bonded protons
It depends on the concentration. In concentrated solutions, these protons are deshielded by hydrogen
bonding. They absorb at a relatively low field. N-H (o 3.5) OH (o 4.5). When it is diluted with a
non-hydrogen-bonding solvent, such as CCl4, hydrogen bonding becomes less important.
Hydrogen bonding and the protons exchange that accompanies it may contribute to a broadening od
the peak from an O-H or N-H proton. A broad peak appears because protons exchange from one
molecule to another during the NMR resonance.
Carboxylic acid protons are bonded to an oxygen next to a carbonyl protons, they have considerable
positive character. Carboxylic acids frequently exist as hydrogen-bonded dimers, with moderate
rates of proton exchange that broaden the absorption of the acid proton. They are strongly deshielded
and absorb at chemical greater than o 10 (normally around o 12).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
207
13.6 The number of signals
In general, the number of NMR signals corresponds to the number of different kinds of protons
present in the molecules (chemically non-equivalent).
Protons in identical chemical environments with the same shielding have the same chemical shift.
Such protons are said to be chemically equivalent.
13.7 Area of the peaks
The area under a peak is proportional to the number of hydrogens contributing to that peak. We
cannot simply compare peak heights. NMR spectrometers have integrators that compute the relative
areas of peak.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
208
13.8 Spin-Spin Splitting
Consider the spectrum of 1,1,2-tribromoethane.
There are two signals with areas in the ratio of 1:2. The splitting of signals into multiplets, called
spin-spin splitting, results when two different types of protons are close enough that their magnetic
fields influence each other. Such protons are said to be magnetically coupled.
The two H
b
protons do not split each other because they are chemically equivalently and absorb at
the same chemical shift.
The N+1 rule:
If a signal is split by N neighboring equivalent protons, it will be split into N+1 peaks.
The relative areas of the N+1 multiplet that results are approximately given by the appropriate line
of Pascals triangle.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
209
The range of magnetic coupling
Most spin-spin splitting is between protons on adjacent carbon atoms
Geminal coupling
Vicinal coupling
Long range coupling
Bonded to nonadjacent carbons: four or more bonds between protons
Leaning of a multiplet. A multiplet often leans upward toward the protons that are causing the
splitting. The ethyl multiplets in ethylbenzene lean toward each other.
H
a
, H
b
, H
c
are chemical nonequivalent.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
210
Coupling constants
The distance between the peaks of a multiplet (measured in hertz) is called the coupling constant.
Coupling constant are represented by J, and the coupling constant between H
a
and H
b
is represented
by J
ab
. A spectrometer operating at 300 MHz records the same coupling constants as a 60 MHz
instrument.
Proton NMR spectrum of 4,4-dimethylcyclohex-2-en-1-one
Problem: which peaks belong to o-hydrogen? |-hydrogen?
Problem-solving hint:
Protons on the |-carbon of an o,|-unsaturated carbonyl compound absorb at very low fields (about o
7) because of the electron-withdrawing resonance effect of the carbonyl group.
More electron density / shielding / up field
Less electron density / deshielding / downfield.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
211
13.9 Complex splitting
Complex organic molecules usually have complex splitting. For example: styrene.
For splitting of H
a
For splitting of H
b
Spectrum of trans-hex-2-enoic acid was shown below. Assign each peaks!!
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
212
13.10 Stereochemical nonequivalence of protons
Stereochemical difference often result in different chemical shifts for the protons on the same carbon.
Consider 3-bromopropene, there four different types of protons in allyl bromide.
If the same product is formed by imaginary replacement of either of two protons, those protons
are chemical equivalent.
Diastereotopic protons
Replace cis hydrogen give cis diastereomers, replace trans hydrogen give trans diastereomers.
Because the two imaginary products are diastereomers, these protons on C1 are called
diastereomeric protons. Diastereotopic protons appear in the NMR at different shifts, and they can
split each other (germinal coupling, H
a
and H
b
).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
213
Diastereomerism also occurs in chiral acyclic molecules. For example 1,2-dichloropropane.
Diastereomeric protons in Newman project.
Real spectrum of 1,2-dichloropropane. Sometimes we called this is a ABX system.
13.11 Time dependence of NMR spectroscopy
A terminal alkynes does not give a spectrum where the molecules oriented along the field absorb at a
high field and those oriented perpendicular to the field absorb at a low field. What we really see is
one signal (o 2.5) whose position is averaged over the chemical shifts of all the orientation of a
rapid tumbling molecules. In general, any types of movement or change that takes place faster
than about a hundredth of a second will produce an averaged NMR spectrum. (like a camera)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
214
Conformational changes
The axial and equatorial hydrogens are inter-convertible through chair-chair interconversion. The
interconversion are fast on an NMR time scale at room temperature. Therefore it shows only a sharp
peak at o 1.4 at r.t. However, the temperature is at 89
o
C, two nonequivalent types of protons that
split each other, giving two borad bands corresponding to the absorptions of the axial and equatorial
protons.
Fast proton transfer
As shown above (Figure-a) , coupling between the hydroxyl (-OH) proton and the adjacent
methylene (-CH2-) protons is about 5 Hz. It is an untrapure sample of ethanol with no contamination
of acid, base, or water. In Figure-b, no splitting is seen between the hydroxyl proton and the
methylene. What we see is a single, unsplit hydroxyl absorption corresponding to the averaged field
the proton experience from bonding to many different ethanol molecules. Proton exchange occurs
in most alcohols and carboxylic acids, and in many amines and amides. If the exchange is fast
(as it usually is for OH group), we see one charp averaged signal. If the exchange is very slow, we
see splitting, if the exchange is moderately slow, we may see a broadened single peak.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
215
Sometimes, it is difficult to tell whether or not a given peak corresponding to one of the
exchangeable protons. To identify which protons are exchangeable protons, we shake the sample
with small amount of deuterium oxide, D2O. Or use CD3OD to take the NMR spectrum. An
exchangeable hydrogens are quickly replaced by deuterium atoms, which are invisible in the proton
NMR spectrum.
Problem-solving strategy
1. Determine the elements of unsaturation.
2. Use typical values of chemical shifts to determine the partial structure
3. Try to connect the chemical bonding to come up with a correct structure.
Determine the structure
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
216
13.12 Carbon-13 NMR spectroscopy
What about if the organic molecules do not contain protons, such as internal alkyne?
Carbon NMR signal are much weaker than proton signals. About 99% of the carbon atoms in a
natural sample are the isotope
12
C.
12
C isotope has an even number of protons and an even number of
neutrons, so it has no magnetic spin and cannot give rise to NMR signals. The less abundant isotope
13C has odd number of neutrons, giving is a magnetic spin of 1/2, just like a proton. Only 1% of the
carbon atoms in a sample are the magnetic 13C isotope, the sensitivity of
13
C NMR is decreased by a
factor of 100.
Fourier Transform NMR spectroscopy
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
217
The precession of many nuclei at slightly different frequencies produces a complex signal that
decays as the nuclei lose the energy they gained from the pluse. This signal is called a free
induction decay (FID) and it contains all the information needed to calculate a spectrum.
A fourier transform is the mathematical technique used to compute the spectrum from the free
induction decay. And this techniques of using pluses and collecting transients is called fourier
trandform spectroscopy.
Carbon chemical shift
Important different between proton and carbon techniques
Operating frequency
The resonance frequency of 13C is about one fourth of proton. A spectrometer is with 70459 guess
magnet needs a 300 MHz transmitter for protons and a 75.6 MHz transmitter for
13
C.
Peak areas
The areas of
13
C NMR peaks are not necessarily proportional to the number of carbons giving
rise to the peaks. Carbon atoms with two or three protons attached usually give the strongest
absorption, and carbons with no protons tend to give weak absorption.
Spin-Spin splitting
As we discussed before, only 1% the carbon atoms in the
13
C NMR sample are magnetic, so there is
only a small probability that an observed
13
C nucleus is adjacent to another
13
C carbon.
Therefore, carbon-carbon-splitting can be ignored. However, carbon-hydrogen coupling is
common. Extensive carbon-hydrogen coupling produce splitting patterns that can be complicated
and difficult to interpret.
To simplify 13C NMR spectrum, they are commonly recorded using proton spin decoupling. Where
the protons continuously irradiated with a broadband proton transmitter. As a resuly, all of the
protons are continuously in resonance, and they rapidly flip their spins. The carbon nuclei see an
average of the possible combination of protons spin states. Each carbon signal appears as a single.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
218
Off resonance decoupled (upper) and broadband decoupled(lower)
13
C NMR spectrua.
DEPT
DEPT (distortionaless enhances polarization transfer) is a technique that provides the same
information as off-resonsnce decoupling. DEPT is easier to run on a modern computer-controlled FT
NMR. Each 13C nucleus is magnetically coupled to the products bonded to it. Under the right
circumstance, this magnetic coupling allows the transfer of polarization from the protons to the
carbon nucleus. The number of protons bondned to the 13C nucleus determines how this polarization
transfer occurs. A DEPT experiment usually includes three spectral scans.
1. A normal decouplied scan
2. A DEPT-90 scan , in which only the CH (methine) carbon bonded to exactly one proton appear
3. A DEPT-135 scan, in which the CH3 (methyl) groups and CH (methane) groups appears
normally, and the CH2 groups give negatives peaks. Carbons that are bonded to no protons do
not appear.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
219
13.13 Interpreting carbon NMR spectra
Same as 1H NMR spectra, carbon spectra are often easier to interpret
1. The number of different signals implies how many different types of carbon are present
2. The chemical shifts of those signals suggest what tyoes of functional groups contain those carbon
atoms.
3. The splitting of signals in the off-resonance decoupled spectrum or DEPT-90 and DEPT-135
spectra indicate how many protons are bonded to each carbon atom.
An impurity of allyl bromide
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
220
During the preparation of 4-hydroxybutanoic acid
Conversion of cyclohexanol to cyclohexyl bromide with large excess of conc. Sulfuric acid
13.14 Nuclear Magnetic Resonance Imaging (MRI)
Medical NMR imaging is commonly called magnetic resonance imaging (MRI) to avoid the
common fear of the word nuclear: and the misconception that nuclear means radioactive.
There is nothing radioactive about an NMR spectrometer. In fact, MRI is the least invasive, least
hazardous method available for imaging the interior of human body. MRI can easily distinguish
watery tissues, fatty tissues, bond, air spaces, bloodetc. It becomes more and more useful in
diagnosis of cancer or tumor.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
221
Chapter 14 Ethers, Epoxides, and thioethers
Symmetric ether and unsymmetric ether
How to make unsymmetric ether?
The most important commercial ether is diethyl ether, often called ethyl ether. Ether was used as a
surgical anesthetic for over a hundred years. Several compounds that are less flammable and more
easily tolerated are now is use, including nitrous oxide (N
2
O) and halothane (CF
3
-CHClBr).
14.2 Physical properties of ethers
Pure ether have no hydroxyl groups to engage in hydrogen bonding, but they can serve as a
hydrogen bonding acceptor with other compounds, which do have O-H bond and N-H bonds.
Ether as polar solvents
Their relatively low boiling points simplify their evaporation from the reaction products.
Polar substances tend to be nearly as soluble in ethers as in alcohols because ethers have large dipole
moments as well as the ability to serveas hydrogen bond acceptors. The nonbonding electron pairs of
an ether effectively solvate cations.
Ether are nonhydroxylic (no hydroxyl group), and they are normally unreactive toward strong bases.
For this reason, ethers are frequently used as solvents for very strong polar bases (like Grignard
reagents) that require polar solvents. The following four ethers shown below are common solvents
for organic reactions. Order in boiling points.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
222
Stable complexes of ethers with reagents
Complexes with electrophiles
Ethers nonbonding electrons could stabilize borane, BH3. Pure borane exists as a dimer called
diborane, B2H6, which is a toxic gas, not easy to handle. Borane forms a stable complex with
tetrahydrofuran, BH3.THF is commercially available as a 1 M solution, easily measured and
transferred like any other air-sensitive liquid reagent. The availability of BH3.THF has contributed
greatly to the convenience of hydroboration.
Boron trifluoride is used as a Lewis acid catalyst. The complex of BF3 with diethyl eterh is called
boron trifluoride etherate
Crown ether complexes
In Chapter 6, we encountered the use of crown ether to solvate the metal cation, in order to increase
nucleophility of the anion (Nuc
). Different crown ethers solvate different cations, depending on the
realtives sizes of the crown ether and the cation and the number of binding sites around the cation.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
223
14.3 Nomenclature of ethers
Common names (alkyl alkyl ether)
Ethers are formed by naming the two alkyl groups on oxygen and adding the word ether, like methyl
tert-butyl ether.
IUPAC names (alkoxy alkane ether)
Use the complex alkyl group as the root name, and the rest of the ether as an alkoxy group.
Nomenclature of cyclic ethers
Epoxides (Oxiranes)
As we have learned in Chapter 8.12. Alkenes reacted with mCPBA gave epxoides.
Draw by yourself!!!
One systematic method for naming epoxide is to name the rest of the molecule and use the
term epoxy as a substituent, giving the numbers of the two carbon atoms bonded to the epoxide
oxygen.
Oxetanes (least common cyclic ether)
They are not as reactive as the highly strained oxiranes, however, they are more reactive than larger
cyclic ether and open chain ethers.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
224
Furans (oxolanes)
The five-membered cyclic ethers are commonly named after an aromatic memner of this group,
furan. The saturated one is called tetrahydrofruan (THF), which is a very common solvent in organic
synthesis. Grignard reactions sometimes succeed in THF even whrn they fail in diethyl ether.
[temperature!!] *Furanose is a name to describe 5-membered ring of carbohydrates
Pyrans (oxanes)
The six-membered cyclic ethers are commonly named as derivatives of pyran, an unsaturated ethers.
With four more hydrogen atoms, it is called tetrahydropyran (THP), an common protective group of
alcohol. * Pyranose is a name to describe 6-membered ring of carbohydrates.
Dioxanes
Heterocyclic ethers with two oxygen atoms in a six-membered ring are called dioxanes. The most
common dioxane is 1,4-dioxanes. 1,4 dioxane is miscible with water, and it is widesly used as a
polar solvents for organic reactions.
Dioxin is a common name for dibenzo-1,4-dioxane, which is 1,4-dioxane fused with two nemzeme
rings. The name dioxin is often used incorrectly in the news media for
2,3,7,8-tetrachlorodibenzodioxin (TCDD). Most dioxins are toxic and carcinogenic (cuase cancer).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
225
14.4 Spectroscopy of ethers
IR
IR spectra do not show obvious and reliable absorption for ethers. Most ethers give a moderate to
strong C-O stretch around 1200 cm
1
(in the fingerprint region). But many compounds other than
ethers give similar absorption. IF the molecules formula contains an oxygen, the lack of carbonyl
(~1715 cm
1
) and hydroxyl absorption (~3300 cm
1
) in the IR suggests an ether.
Mass spectrometry
The most common fragmentation of ethers is alpha cleavage (o cleavage). It is cleavage next to the
carbon bonded to oxygen. The resulting oxonium ion is resonance-stabilized by the nonbonding
electrons on oxygen.
NMR
1H NMR the peaks of H-C-O is around 3-4 ppm
13C NMR the peaks of C-O is around 55-90 ppm
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
226
14.5 The Williamson ether synthesis
As we already discussed in section 11-14.
What you need to remember is it always follows the S
N
2 attack of an alkoxide on an
unhindered primary alkyl halide or tosylate. Elimination may compete with the use of a
secondary halide, poor yield often obtained. Draw a reaction equation below by yourself.
14.6 Synthesis of ethers by alkoxymercuration-demercuration
As we already discussed in section 8.6. Remember it follow the Markovnikovs rule. Draw a
reaction equation below by yourself.
14.7 Industrial synthesis of biomolecular condensation of alcohols
The least expensive method for synthesizing simple symmetric ethers is the acid-catalyzed
bimolecular condensation, which we already discussed in section 11.10. Condensation means
joining of two molecules, with loss of a small molecule, like water). Draw a reaction equation
below by yourself.
* Remember that it is only used for the synthesis of symmetric ethers. Unsymmetric ethers have to
be prepared by Williamson ether synthesis.
* Should this condensation reaction be carried out in a relatively high (180
o
C) or relatively low
(140
o
C) temperature?
14.8 Cleavage of ethers by HBr and HI
Although ethers are commonly used as organic solvents, it does undergo a limited number of
characteristic reactions. Under the acidic conditions, ethers are cleaved by heating with HBr or HI to
give alkyl bromides or alkyl iodides. The conditions are very strong, however, and the molecule
must not contain any acid-sensitive functional groups.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
227
14.9 Autoxidation of ethers
When ethers are stored in the presence of atmospheric oxygen, they slowly oxidize to produce
hydroperoxides and dialkyl peroxides, both of which are explosive. Such a spontaneous oxidation by
atmospheric oxygen is called autoxidation. Distillation of ethers may cause explosive due to ether
peroxides
14.10 Thioethers (sulfides) and silyl ethers
Thioethes also claaed sulfides, are ethers with a sulfur atom replacing the oxygen atom of an ether.
Silyl ethers are ethers with a substituted silicon atom replacing one of the alkyl groups of an ether.
Silyl ethers share some of the properties of ethers (resistant to some acids, bases, and oxidizing
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
228
agents), but they are more easily formed and more easily hydrolyzed. These properties make them
useful as protecting groups, and silyl ethers are frequently used to protect alcohols.
Thioethers (sulfides)
Like thiols, thioethers have strong characteristic odors. Sulfides are named like ethers, with sulfide
replacing ether in the common names. In the IUPAC (alkoxy alkane) names, alkylthio replaces
alkoxy
Thiols are more acidic than water. Therefore, thiolate ions are easily generated by treating thiols with
aqueous sodium hydroxide
Because sulfur is larger and more polarizable than oxygen, thiolate ions are even better nucleophiles
than alkoxide ions. Thioethers are easily synthesized by the Williamson ether synthesis, using a
thiolate ion as the nucleophile. Remember it goes through S
N
2 reaction mechanism.
Sulfides are much more reactive than ethers. Sulfur can from additional bonds with other atoms.
Sulfides are easily oxidized to sulfoxides and sulfones. Sulfoxides and sulfones are draw using
either hypervalent double-bonded structures or formally charged single-bonded structures as shown
here.
As you might remember that dimethyl sulfide are often used as mild reducing agents for ozonolysis
of alkanes.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
229
Sulfur compounds are more nucleophilic than the corresponding oxygen compounds. Because sulfur
is larger and more polarizable and its electrons are less tightly held in orbitals that are farther from
the nucleus. Sulfides attack unhindered alkyl halides to give sulfonium salts. Sulfonium salts are
strong alkylating agents because the leaving group is an uncharged sulfide.
Silyl ethers as alcohol-protecting groups (Organic Synthesis)
Synthetic chemists have developed many different silyl protecting groups that vary widely in their
reactivity and are carefully chosen for a specific use. In aqueous or organic solvents, fluoride ion
removes silyl ethers under gentle conditions because the silicon-fluoride bond is exceptionally strong.
(The Si-F bond is the strongest covalent bond)
Requirements for a protecting group
1. Easy to put in
2. Resistant to reagents that would react with unprotected functional groups.
3. Easy to remove
Protection of alcohols
We have mentioned the THP groups as alcohol protecting group, but by far the most popular groups
for alcohols are the various silyl groups. Consider O-Si bond energy Draw the following
structures by yourself!!!
TMS (trimethylsilyl) easy to fall off.
TES (triethylsilyl)
TIPS (tri-iso-propylsilyl)
TBDMS (TBS, tert-butyl-dimethylsilyl) t-BuMe
2
Si-
TBDPS (tert-butyl-diphenylsilyl) t-BuPh
2
Si-
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
230
14.11 Synthesis of epoxides
Peroxyacid epoxidation
As we mentioned in section 8.12, epoxides could be made from alkenes under the mCPBA
epoxidation. Meta-chloroperoxybenzoic acid (mCBPA) is often used for these transformations.
Draw an equation below by yourself!!
Theperoxyacid epoxidation is quite general, with electron-rich double bonds reacting fastest. The
second example uses magnesium monoperoxyphthalate (MMPP), a relatively stable water-soluble
peroxyacid often used in large-scale epoxidation. These aqueous MMPP epoxidations, carried out at
neutral pH to avoid opening the epoxide, avoid the larg-scale use of hazardous chlorinated solvents.
Base-promoted cyclization of halohydrins
Another method to make epoxide anmd other cyclic ethers involving an intramolecular Williamson
ether synthesis. If an alkoxide ion and a halogen atom are located in the same molecule,
base-catalzed cyclization takes place to form epoxide or cyclic ethers, Treatment of a halohydrin
with a base leads to the formation of an epoxide through an intramolecular S
N
2 attack.
Step 1: formation of halohydrin form alkene
Step 2: base-mediated cyclization
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
231
This reaction can be used to synthesize cyclic ether with larger rings. The difficulty lies in
preventing the base (added to deprotonate the alcohol) from attacking and displacing the halides.
2,6-Lutidine, a bulky base that cannot easily attack a carbon atom, can protonate the hydroxyl group
to give a five-membered cyclic ether. 5-, 6-, 7-membered cyclic ethers are obtained in this way.
Could you have the ability to tell the advantages and disadvantages of these two epoxidation
methods? Hint: give them some structures with special functional groups!
14.12 Acid-catalyzed ring opening of epoxides
Epoxides are much more reactive than dialkyl ethers because of the ring strain energy (25 kcal/mol)
associated with the three-membered ting.
In water
In section 8.13, we saw acid-catalyzed hydrolysis of epoxides gives glycols with anti-diol
stereochemistry.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
232
Do you remember how to make syn-diol from an alkene?
In alcohol
What about if the reaction carry out in alcohol? Draw an equation here by yourself!
Using hydrohalic acids
This reaction is analogous to the cleavage of ethers by HBr and HI to give 1,2-dihalides. However,
you could make it by electrophilic addition of X
2
.
14.13 Base-catalyzed ring opening of epoxides
Most ether do not undergo nucleophilic substitutions or elimination under basic condensation,
because an alkoxide ion is a poor leaving group. However, base-catalyzed ring opening of the
epoxide is possible due to the 3-membered ring strain.
14.14 Orientation of epoxide ring opening
An unsymmetric epoxide may produce different product under acid-catalyzed and base-catalyzed
conditions. You need to remember!!!
In acid-catalyzed ring opening
Protonated epoxides cause slightly more positive charge (o
+
) on the more-substituted carbon. It will
guide the nucleophiled to attack the carbon with more positive charge (o
+
).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
233
In base-catalyzed ring opening
No protonation at all. Bases (nucleophiles) would like to attack the less hindered carbon (less
substituted carbon) to delivery different product.
14.15 Reactions of epoxide with Grignard and organolithium reagents
This section is so simple.
Do Grignard reagents and organolithium reagents serve as bases or acids?
It usually attacks the less hindered carbon.
14.16 epoxy resin: the advent of modern glues
Epoxide polymerize in place, so they match the shape of the joint perfectly and adhere to
microscopic irregularities in the surfaces. There is no solvent to evaporate, so there is no shrinkage.
Epoxides are bonded by ether linkages, so they are unaffected by water. The most common epoxy
resins use a prepolymer made from bisphenol A and epichlorohydrin.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
234
Chapter 15 Conjugated systems, orbital symmetry, and ultraviolet
spectroscopy
Isolated double bond / conjugated double bond / cumulated double bond
15.2 Stabilityes of dienes
Because penta-1,2-diene has a larger heat of hydrogenation than penta-1,4-diene, we conclude that
the cumulated double bonds of allenes are less stable than isolated double bonds, and much less
stable than conjugated double bonds.
15.3 Molecular orbital picture of a conjugated system
As the figure shown above, the compound with conjugated double bonds is 17 kJ /mol (4.0 kcal/mol)
more stable than a similar compound with isolated double bonds. This 4 kcal/mol of extra energy in
the congujated molecule is called the resonance energy. Or called conjugated energy,
delocalizationh energy, and stabilization energy.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
235
The bond between two double bonds is about 1.48 , which is shorter than a normal carbon-carbon
single bond. This bond is shortened slightly by the ioncreased s character of the sp2 hybride orbitals,
but the most important cause of this short bond is it pi bonding overlap and partial double-bond
character. In fact, the electrons in the double bonds are delocalized over the entire molecule.
Molecular orbitals
You have learned this when you studied General Chemistry.
Molecular orbitals of buta-1,3-diene
Butadiene has four pi electrons (two electrons in each of the two double bonds in the Lewis structure)
to be placed in four MOs. Each MO can accommodate two electrons, and the lowest-energy MOs are
filled first. Therefore, the four pi electrons go into t
1
and t
2
. Both bonding MOs are filled, and both
antibonding MOs are empty. Most stable molecules have this arrangement of filled bonding orbitals
and vacant antibonding orbitals. The following figure also compares the relative energies of the
ethylene MOs with the butadiene MOs to show that the conjugated butadiene system is slightly more
stable than two ethylene double bonds.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
236
The partial double-bond character between C2 and C3 in buta-1,3-diene explains why the
molecule is most stable in a planar conformation. There are actually two planar conformations
that allow overlap between C2 and C3. These conformations arise by rotation about the C2-C3 bond,
and they are considered single-bond analogues of trans and cis isomers about a double bond. Thus,
they are named s-trans (single-trans) and s-cis (single-cis) conformations.
The s-trans conformation is 12 KJ /mol (2.8 kcal/mol) more stable than s-cis conformation, which
shows interference between the two nearby hydrogen atoms. The barrier for rotation about C2-C3
bond from s-trans to s-cis is only about 29kJ /mol (about 7 kcal/mol). The s-cis and s-trans
conformers of butadiene easily interconvert at room temperature.
15.4 Allylic cations / 15.7 Allylic radicals
Rebebmer I have you to memorize the bond dissociated energy of allylic C-H bond and benzylic
C-H bond?
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
237
Allylic cation
Also remember this chemical transformation? It involved the formation of allylic cation.
Stability of carbocations
Allylic radical
Bromination through allylic radical
H H
Br
2
, or NBS
hv
Br H
Reaction mechanism
Step 1: generation of a bromo radical
Step 2: through resonance-stabilization allylic radical
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
238
Propose a reaction mechanism for the following transformation?
Mechanism:
15.5 1,2- and 1,4-addition to conjugated dienes
You have learned the Markovnikovs product of electrophilic addition of alkenes with HBr.
What about this, electrophilic addition of conjugated diene with HBr? Any others products would
be obtained?
Answer: 1,2- and 1,4-addition products. Draw by yourself.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
239
15.6 Kinetic versus thermodynamic control in the addition of HBr to
buta-1,3-diene
One of the interesting peculiaries in the reaction of buta-1,3-diene with HBr is the effect of
temperature on the products, as below.
Considering the delocalized allylic cation (resonance-stabilized allylic cation), could you determine
which reaction intermediate is more stable (the energy state is much lower)? The allylic cation with
secondary carbon [(1,2)
+
], or the allylic cation with primary carbon [(1,4)
+
]?
Reaction-energy diagram
Kinetic control at 80
o
C
1,2-addition results from bromide attack at the more substituted secondary carbon, which bears
more of the positive charge because it is better stabilized than the primary carbon. Because the
1,2-addition has a lower activation energy (low Ea) than the 1,4-addition, the 1,2-addition takes
place faster (at all temperature)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
240
The product that is formed faster predominates. Because the kinetics of the reaction determine the
results, this situation is called kinetic control of the reaction. The 1,2-product, favored under these
conditions, is called the kinetic product.
Thermodynamic control at 40
o
C
At 40
o
C, a significant fraction of molecular collisions have energy for reverse reactions to occur.
At this point, an equilibrium is set up, and the relative energy of the each species determine its
concentration. The 1,4-addition is the most stable species, and it predominates. Since the
thermodynamics determine the results, this situation is called thermodynamic control (or
equilibrium control) of the reaction. The 1,4-addition, favored under these conditions, is called the
thermodynamic product.
The key point:
In general, reactions that do not reverse easily are kinetically controlled because no equilibrium is
established. In kinetic controlled reactions, the product with the lowest energy transition state
predominates. Reactions that are easily reversible are thermodynamically controlled. In
thermodynamically controlled reactions, the lowest-energy product predominates.
15.8 Molecular orbitals of the allylic system / 15.9 Electronic configuration of the
allyl radical, cation, and anion
With an odd number of MOs, they cannot be symmetrically divided. One of the MO must appear at
the middle of the energy levels, either bonding nor anitbonding: It is nonbonding molecular orbital.
Electrons in a nonbonding orbital have the same energy as in an isolated p orbital.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
241
In the allyl radical, it lacks the unpaired electron in t
2
, which has half of its electron density on C1
and half on C3. This MO picture is consistent with the resonance picture showing the positive charge
shared by C1 and C3.
In allyl anion, MO presents a negative charge and a lone pair of nonbinding electrons evenly divided
between C1 and C3.
15.10 S
N
2 displacement reactions of allylic halides and tosylates
Allylic halides and tosylates show enhanced reactivity toward nucleophilic displacement reactions
by the S
N
2 mechanism. Allyl bromide reacts with nucleophiles by the S
N
2 mechanism about 40
times faster than n-propyl bromide. When the substrate is allylic, the transition state receives
resonance stabilization through conjugation with the p orbitals of the pi bond. This stabilization
lowers the energy of the transition state, resulting in a lower activation energy and an enhanced rate.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
242
15.11 The Diels-Alder Reaction
German chemists Otto Diels (Kiel University) and Kurt Alder (Kln University) discovered this
useful and powerful reaction. The alkenes and alkynes wit electron-withdrawing groups add to
conjugated dienes to form six-membered rings. They were awarded the Nobel Prize in 1950 for their
contribution.
CN
heat
CN
diene dienophile
(4 electrons) (2 electrons)
Diels-Alder product
The Diels-Alder reaction:
The Diels-Alder reaction is called a [4+2] cycloaddition because a ring is formed by the interaction
of four pi electrons in diene with two pi electrons of the alkene or alkyne. The electron-poor
alkene and alkyne is prone to react with diene, it is called a dienphile (love of dienes). In fact, the
Diels-Alder convert two pi bonds into two sigma bonds. This electron movement is concerted,
with three pairs of electrons moving simultaneously.
The diene is electron-rich and serving as a nucleophile. The presence of electron-donating (-D)
groups, such as the alkyl groups or alkoxy (-OR) groups, may further enhance the reactivity of the
diene. The dienophile is electron-poor and serving as an electrophile. A good dienophile generally
has one or more electron-withdrawing (-W) pulling electron density away from the pi bond.
Dienophiles commonly have carbonyl-containing (C=O) groups or cyano (-CN) groups to enhance
their Diels-Alder reactivity. Some examples are shown below.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
243
A: Stereochemical requirement of the Diels-Alder Transition state.
The Diels-Alder reaction is a concerted cyclic movement of six electrons: four in the diene and two
in the dienophile. For the three pairs of electrons to move simultaneously, the transition state must
have a geometry that allow overlap of the two end p orbital of the diene with those of the
dienophile, as shown below.
B: s-Cis conformation of the diene
As you have learned the geometry of s-trans and s-cis. Would you expect that which one will be
suitable for Diels-Alder reaction.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
244
Structural feathers that aid and hinder the diene in achieving the s-cis conformation affect its ability
to participate in Diels-Alder reactions. More likely to be s-cis conformation, the faster is the reaction
rate. Dienes that easily adopt the s-cis conformation undergo the Diels-Alder reaction more
readily.
The faster one in the above arrow is cyclopentadiene. Bacuase cyclopentadiene is fixed in the s-cis
conformation It is highly reactive in Diels-Alder reaction. It even dimerize to form
dicyclopentadiene at room temperature. Monomer could be obtained by heating the dimer above
200
o
C. The monomer could be stored at dry-ice temperature or distilled from dimerization before
use.
C: Syn stereochemistry
The Diels-Alder reaction is a syn addition wit respect to both the diene and dienophile. The
dienophile adds to one face of the diene, and the diene adds to once face of the dienophile. The
following examples show the results of this syn addition.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
245
D: The endo rule
When the dienophile has a pi bond in its electron-withdrawing group (as in a carbonyl group or a
cyano group), the p orbitals in that electron-withdrawing group approach one of the central carbon
atoms (C2 or C3) of diene. This proximity results in secondary overlap: an overlap of the p orbitals
of the electron-withdrawing group with the p orbitals of C2 and C3 of the dene. Secondary overlap
helps to stabilize the transition state.
In the bicyclic product (called norbornene), the electron-withdrawing substitutent occupies the
stereochemical position closet to the central atoms of the diene. This position is called the endo
position because the substituent seems to be inside the pocket formed by the six-membered ring of
norbornen. This stereochemical preference of the electron-withdrawing substituent to appear in the
endo position is called the endo rule.
The endo rule is useful for predicting the products of many types of Diels-Alder reaction. Try to do
some exercises.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
246
Are you able to predict the endo product is kinetic product or thermodynamic product?
Which transition state has lower activation energy?
Which product has more steric hinderence?
Using unsymmetric reagents
If the diene and dienophile have substituents, what products will you expect to obtain?
Electro-donating group (-D) on diene, electron-withdrawing group on dienophile, what are their
relationship in positions?
Although it is a concerted mechanism, to understand this results, the charge-separated resonance
forms will help us to remember this outcome. If there is a donating group at C2 position of diene,
through charge-separated resonance forms, anionic C1 carbon will like to undergo conjugated
addition (1,4-addition) to the dienophile and form 1,4- Diels-Alder product, as shown below.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
247
However, if there is a electron-donating group at C1 of the diene, the anionic C4 carbon will like to
undergo conjugated addition (1,4-addition) to the dienophile and form 1,2- Diels-Alder product, as
shown below.
In Most cases, we do not need to draw the charge-separated resonance forms to determine the result,
We will remember 1,2- or 1,4-product. The electron-donating group (-D) of the diene and
electrons-withdrawing group (-W) of the dienopile usually bear either a 1,2-relationship or 1
1,4-relationship, no 1,3-relationship will be observed. Another way is to remember is ortho-, or
para- position if it will finally become a benzene ring.
15.12 The Diels-Alder as an example of a pericyclic reaction
Thye Diels-Alder is one class of concerted pericyclic reactions. A concerted pericyclic reaction has a
single transition state, whose activation energy may be supplied by heat (thermal induction) or by
ultraviolet light (photochemical). Some pericyclic reactions proceed only under thermal condition,
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
248
and others proceed only under photochemical induction. Some pricyclic reactions take ploace under
both thermal and photochemical condition, but the two sets of conditions give two different
products.
In 1965, Robert B. Woodward (1965 Nobel Prize) and Ronald Hoffmann (1981 Nobel Prize)
developed a theory for predicting the results of pericyclic reactions by considering the symmetry of
the molecular orbitals of the reactants and products. This theory, called conservation of orbital
symmetry, Woodward-Hoffmann rules. It says that the molecular orbitals of the reactants must
flow smoothly into the MOs of the products without any drastic changes in symmetry, In that case,
there will be bonding interactions to help stabilize the transition state.
The electrons in the highest-energy occupied orbital, called the Highest Occupied Molecular
Orbital (HOMO). The orbital in ethylene that receives these electrons is the lowest-energy orbital
available, the Lowest Unoccupied Molecular Orbital (LUMO). If the electrons in the HOMO of
butadiene can flow smoothly into the LUMO of ethylene, a converted reaction (Diels-Alder
reaction) can take place. In the figure shown above, the HOMO of butadiene has the correct
symmetry to overlap in phase with the LUMO of ethylene. Having the correct symmetry means
the orbitals that form the new bonds can overlap constructively: plus with plus, and minus
with minus. The bonding interactions stabilize the transition state and promote the concerted
reaction. This favorable result predicts that the reaction is symmetry-allowed.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
249
The thermal forbidden [2+2] cycloaddition
The [2+2] cycloaddition requires the HOMO of one of the ethylene to overlap with the LUMO of the
other. However, if a cycloaddition produces an overlap of the positive-phase orbitals with
negative-phase, antibonding are generated, which will raise the activation energy, so the reaction is
classified as symmetry-forbidden. The reaction wont take place in thermal conditions.
However, [2+2] cycloaddition reaction is photochemically allowed. When ultraviolet (UV) light is
used, one of the p electrons ix excited to the next higher molecular orbital. This higher orbital,
formerly the LUMO, is now occupied. IT is the new HOMO*. The HOMO* of the excited ethylene
molecule has the same symmetry as LUMO of a grouind-state ethylene. Any excited molecule can
react with a ground-state molecule to give cyclobutane.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
250
The [2+2] cycloaddition is therefore photochemically allowed but thermally forbidden. In general,
photochemically allowed reactions are thermally forbidden, and thermally allowed reactions
are photochemically forbidden.
15.13 Ultraviolet absorption spectroscopy
Untraviolet (UV) spectroscopy is used to detects the electronic transitions of conjugated systems and
provides information about the length and structure of the conjugated part of a molecule.
Spectral region
Ultraviolet frequencies correspond to shorter wavelengths and much higher energies than visible and
infrared. UV operate in the range of 200 to 400 nm, corresponding to photo energies of about 300 to
600 nm. These spectrometers often extend into the visible region and are called UV-visible
spectrometers.
Ultraviolet light and electronic transitions
The wavelengths of UV light absorbed by a molecule are determined by the electronic energy
difference between orbitals in the molecule. Pi bonds have e;ectrons that are more easily excited into
higher energy orbitals. A photo with the right amount of energy can excite an electron from bonding
orbital (t, HOMO) to the anitbonding (t*, LUMO). This transition from a t bonding orbital to a t*
antibonding orbital is called a t t* transition.
The t t* transition of ethylene requires absorption of light is at 171 nm. The t
2
t
3
* transition
of butadiene absorption of light is at 217 nm, while the t
3
t
4
* transition of 1,3,5-hexatriene
absorption of light is at 258 nm. Therefore, we can summarize the effects of conjugation on the
wavelength. A compound that contains a longer chain of conjugated double bonds absorbs light at a
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
251
longer wavelength. The longer the congujation, the longer the wavelength.
For example:
4 conjugation is around 290nm, 5 congujation is around 334,
Isolated double bonds do not contribute to shifting the UV absorption to longer wavelength. Both
their reactions and their UV absorption are like those of simple alkenes.
Ultraviolet spectrometer and ultraviolet spectrum
An ultraviolet spectrometer
The sample is placed in a quartz cell, and some of the solvent is placed in a reference cell. The
spectrometer has a source that emits all frequencies of UV light (above 200 nm). This light passes
through a monochromator, which used a diffraction grating or a prism to spread the light into a
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
252
spectrum and select one wavelength. This light is split into two beams with one passing the sample
cell, the other passing through the reference cell. The detector measures the intensity ratio of the
reference beam (I
r
) compared with the sample beam (I
s
)
The absorption, A, of the sample at a particular wavelength is governed by Beers law.
c =the molar absorptivity ( or molar extinction coefficient) of the sample
c =sample concentration is moles per liter
l =path length of light through the cell in centimeters
The UV spectrum of isoprene is shown below. The concentration is 4 x 10
5 M and the cell is 1 cm.
The information obtained from this spectrum are
max
=222 nm, A =0.8, what is c?
With the c value, you make a curve (A vis concentration)! With this standard curve, you can know
the concentration in any other solution.
Interpreting UV-Vis spectra
We could use some simple generalization for estimating approximate values of
max
for common
types of systems. Here are some good rules: An additional conjugated C=C increase
max
about
30 ~ 40 nm (35 nm); an additional alkyl group increase it about 5 nm. Useful base values.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
253
For organic chemists, although UV spectrum is not as useful as before, but some representative
molecules and some good rules you still need to remember!!
15.14 Colored organic compounds
|-carotene.etc.
15.15 UV-visible analysis in biology and medicine
An example of using UV spectrometer to measure the enzyme inhibition.
If a compound serves as potent enzyme inhibitor, it will block the enzyme active site. Therefore, the
above chemical reaction will not take place. The yellow solution will not show up. While a
non-active molecule is used for this enzyme inhibition test, the yellow solution will show up.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
254
Chapter 16 Aromatic compounds
Aromatic compounds: low hydrogen to carbon ratios; pleasant orders.
Aliphatic compound: fatlike;
16.1/16.2 Discovery, structure and properties of benzene
In 1825, Michael Faraday isolated a pure compound with H:C atom ratio to 1:1. The empirical
formula is (CH)
n
. The structures shown below are the structures of benzene predicted in the past. In
1834, Eilhard Mitscherlich used a vapor-density measurement to determine the molecular weight is
78, for a molecular formula of C
6
H
6
.
Until 1866, Friedrich Kekul proposed a cyclic structure for benzene with three double bonds and
three single bonds. If there are some substituents on the benzene ring, how to tell the difference.
Kekul suggested that a fast equilibrium interconverts the two isomers of benzene, as following.
The resonance representation
In modern time, the spectroscopic methods told us that the benzene is planar and all bonds are the
same length (1.397 ). The carbon nuclei are positioned at equal distance, while two Kekul
structures must differ only in the positioning of the pi electrons. Benzene is actually a resonance
hydride of the two Kekul structures.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
255
Using this respnance picture, we can draw a more realistic representation of benzene. It is a ring of
six sp
2
hydride carbon atoms. Each sp2 carbon atom has an unhybridized p orbital perpendicular to
the plane of the ring, and six electrons occupy this circle of p orbital.
The unusual stability of benzene
By comparing molar heats of hydrogenation of cyclohexene, cyclohexadienes and benzene, we are
able to get a quantitative idea of its stability. Through the experiments, we can calculate the
resonance energy of benzene, which is about 151 kJ /mol (~36 kcal/mol).
Because of this resonance energy, benzene undergoes quite different (unusual) reaction, as compared
to normal alkenes compounds. For example: alkenes will undergo di-hydroxylation reactions with
potassium permanganate to givesyn-diol. However, benzene is inert for this reaction conditions.
Alkenes will undergo bromination with bromine to decolorize the solutions of bromine, however,
benzene is not.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
256
To give a brominated benzene, a catalyst, such as ferric bromide is used to achieve this
transformation. Actually, addition of Br
2
does not take place. Instead, the compound obtained from
substitution of a bromine atom for a hydrogen is formed.
Failures of the resonance picture
The cyclic hydrocarbons with alternating single and double bonds are called annulenes. For
example, benzene is the six-membered annulene, so it can be named [6]annulene. Cyclobutadiene is
[4]annulene, cyclooctatatraene is [8]annulene.
For the double bonds to be completely conjugated, the annulene msut be planar so the p orbitals of
the pi bonds can overlap. As long as an annulene is assumed to be planar, we can draw Kukel-like
structures that seem to show a benzene-like resonance, as following
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
257
Although these resonance structures suggest that the [4] and [8]annulenes should be unusually stable
(like benzene), experiments have shown that cyclobutadiene and cyclooctatatraene are NOT
unusual stable. Cyclobutadiene has never been isolated and purified. It undergoes an extremely fast
Dield-Alder dimerization. To avoid the Diels-Alder reaction, cyclobutadiene has been prepared at
low concentration in the gas phase and as individual molecules trapped in frozen argon at low
temperatures. This is not the behavior we expect from a molecule with exceptional stability.
Richard Willster synthesized cyclooctatetraene in 1911and found it reacts like a normal polymer. In
fact, structural studies have shown cyclooctatetraene is not planar. It is most stable in a tub
conformation, with poor overlap between pi bonds.
Hint:
The molecules with (4n+2) pi electrons are stable (with resonance energy), while those with (4n)
pi electrons are not stable but reactive, as compared with benzene.
~We will discuss a few pages later~
16.3 The molecular orbitals of benzene
We have learned the molecular orbitals of acyclic alkenes, such as ethylene, butadiene. How to
extend this concept to a benzene ring?
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
258
1. Six p orbitals that overlap to form the benzene pi system. Finally give six molecular orbitals
2. The lowest-energy molecular orbital is entirely bonding, with constructively overlap between all
pairs of adjacent p orbital. There are no vertical nodes in this lowest-lying MO.
3. The nuber of nodes increases as the MOs increase in energy
4. The MOs should be evenly divided between bonding and antibonding MOs/
5. A stable system will have filled bonding MOs and empty antibonding MOs.
In a cyclic system of overlapping p orbitals, the intermediate energy levels are degenerate (equal in
energy), with two orbitals at each energy level. Both t
2
and t
3
have one nodal plane.
The energy diagram of benzene
They are symmetrically distributed above and below the nonbonding line. Six electrons fill the three
bonding MOs of the benzene system. This electronic configuration explains the unusual stability of
benzene. The first MO is all-bonding and is particularly stable. The second and third (degenerate)
MOs are still strongly bonding. This configuration, with all the bonding MOs filled (a closed
bonding shell), is energetically very stable.
16.4 The molecular orbital picture of cyclobutadiene
Not like benzene has resonance energy,experimental evidence shows cyclobutadiene is unstable. Its
instability is explained by the molecular orbitals. The lowest-energy MO is t
1
, the all-bonding MO
with no nodes. The next two orbitals, t
2
and t
3
, are degenerate (equal energy), each having one
symmetrically situated nodal plane. The final MO, t
4
*, has two nodal planes and is entirely
anitbonding.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
259
Two electrons fill t
1
, Once t
1
is filled. If others two electrons go into the same orbital, they must
have paired spins and they must share the same region of space. Since electrons repel each other, less
energy is required for the electrons to occupy different degenerate orbitals, with unpaired spins. This
is another application of Hunds rule. The electronic configuration indicates that cyclobutadiene
should be unstable. Its electrons in HOMO are in nonbonding orbital t
2
and t
3
are therefore very
reactive.
The Polygon Rule
The lowest-lying MO is the unique one with no nodes. Thereafter, the molecular orbitals occur in
degenerate (equal-energy) pairs until only one highest-lying MO remains, as below.
The polygon rule states that molecular orbital energy diadram of a regular completely conjugated
cyclic system has the same polygonal shape as the compound, with one vertex (the all-bonding MO)
at the bottom. The nonbonding line cuts horizontally through the center of the polygon. The pi
elelctrons are filled into the orbitals in accordance with aufbau principle (lowest-energy orbitals are
filled first) and the Hunds rule.
16.5 Armoatic, antiaromatic, and nonaromatic compounds
So far you know, some aunnulenes are quite stable with large resonance energy, like benzene. But
some are not, like cyclooctatetraene. Now, we can categorize these molecules.
Aromatic compound
The compounds meet the following criteria
1. Cyclic compounds with some number of conjugated pi bonds
2. Each atom in the ring must have an unhybrided p orbtail. (The ring atmoms are usually sp
2
hybrided or occasionally sp hybridized.
3. The unhybrided p orbitals must overlap to form a continuous ring of parallel orbitals. In most
cases, the structure must be planar for effective overlap.
4. Delocalization of the pi electrons over the ring must lower the electronic energy.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
260
Aromatic structures are mor stable than their open-chain counterparts. For example, benzene is more
stable than hexa-1,3,5-triene.
Antiaromatic compounds
The compounds with the first three criteria, but delocalization of the pi electrons over th ring
increases the electronic energy. Like cyclobutadiene meets the first three criteria for a continuous
ring of overlapping p orbitals, but delocalization of the pi electrons increases the electronic energy.
Cyclobutadiene is less stable than its open-chain counterpart (buta-1,3-diene), and it is antiaromatic.
Nonaromatic compounds
A cyclic compound that does not have a continuous, overlapping ring of p orbitals cannot be
aromatic or antiaromatic. It is said to be nonaromatic. Or aliphatic. Its electronic energy is similar to
that of its open-chain counterpart.
16.6 Hckels rule
A shortcut was developed by Erich Hckel for prediction of these categories:
1. To qualify as aromatic or antiaromatic, a cyclic compound must have a continuous ring of
overlapping p orbitals, usually in a planar conformation.
2. Once it meets the first one, if the number of pi electrons in the cyclic system is:
(4n+2), the system is aromatic.
(4n), the system is antiaromatic. Where n is an integer, commonly 0, 1, 2, or 3.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
261
[4] Cyclobutadiene ([4]annulene) has a continuous ring of overlapping p orbitals. But is has 4N pi
electrons. Hckels rule predicts cyclobutadiene to be antiaromatic.
[6] Benzene has a continuous ring of overlapping p orbitals. And it has 4N+2 pi electrons. It is
aromatic.
[8] Although cyclooxtatetraene has 4N pi electrons, but the p orbitals overlapping is poor. It is
neither antiaromatic nor aromatic. Its reactions are typical of alkenes, as nonaromatic compound.
[10] Aromaticity in the larger (4N+2) annulenes depends on whether the molecule can adopt the
necessary planar conformation. In the all-cis[10]annulene, the planar conformation requires an
excessive amount of angle strain. The [10]annulene isomer with two trans double bonds cannot
adopt a planar conformation either, because two hydrogen atoms interfere with each other.
Neither of these [10]annulene isomer is aromatic, even though each has (4N+2) pi electrons.
When these hydrogen atoms are replaced with a bond (two eclipsed hydrogen atoms were
removed), the compound (naphthalene) is aromatic.
Large-ring annulenes
Like cyclooctatetraene, larger annulenes with (4N) systems do not show antiaromaticity because
they have the flexibility to adopt nonplanar condormations. Even though [12]annulen, [16]annulene,
and [20]annulene are 4N systems. They all react as partially conjugated polyenes.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
262
Some of the larger annulene with (4N+2) pi electrons can achieve planar conformation. For example,
[14]annulene and [18]annulene have aromatic properties.
16.8 Aromatic ions
Hckels rules also applies to systems having odd numbers of carbon atoms and bearing positive or
negative charges. For example: five p orbitals with six pi electrons and 7 p orbitals with six pi
electrons. Are they aromatic? Remember that Hckels rules do not mention anything about atoms, it
only mentions p orbitals and pi electrons.
The cyclopentadienyl ions
With a cyclic compound having five p orbitals, with four pi electrons, it is predicted to be
antiaromatic. With six pi electrons, it is predicted to be aromatic. The MOs energy diagram and p
orbitals are shown in the following.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
263
Because the cyclopentadienyl anion (6 pi electrons) is aromatic, it is usually stable compared with
other carbanions. It can be formed by abstracting a proton from cyclopentadiene. Cyclopentadiene
has a pKa of 16 (almost like water), compared with a pKa of 46 for clcyohexene. It is entirely
ionized by potassium tert-butoxide. Deprotonation of the CH
2
- groups leaves an orbital occupied by
a pair of electrons. This orbital can rehybride to a p orbital, completing a ring of p orbitals containing
six pi electrons. Although it is aromatic, this does not necessarily imply that it is as stable as
benzene.
Hckels rules predicts that the cyclopentadienyl cation (with 4 pi electrons) is antiaromatic. In
agreement with this prediction, it is not easily made. Since neutral antiaromatic compound
(cyclobutadiene) is not easy to isolate, the antiaromatic cation (cyclopentadienyl cation) is simply
too unstable,
Careful:
Even you could draw many resonance forms of antiaromatic compunds, like cyclopentadienyl
cation, that does mean it is a stable compound.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
264
The cycloheptatrienyl ions
What about seven p orbitals with 6 pi electrons or 8 pi electrons?
The cycloheptatrienyl cation (tropylium ion) is easily made by treating the corresponding alcohol
with dilute aqueous sulfuric acid, which means it belongs to aromatic by means of prediction of
Hckels rules. Nevertheless, the tropylium ion is not necessarily as stable as benzene. It only means
this ion is more stable than the corresponding open-chain ion.
The cycloheptatrienyl anion is antiaromatic (4N pi electrons, if it is planar) in agreement with
their behavior in reaction. It is very reactive and not easily made.
The cyclooctatetraene dianion
Although dianion of hydrocarbons are rear and are usually much more difficult to make. The
cyclooctatetraene dianion has planar, regular octagonal structure with C-C bond length of 1.40 ,
and ten pi electrons. It is easily made because it is aromatic.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
265
Summary of annulenes and their ions
Careful:
Some structures with 4N+2 electrons, but not planar
Some structures like 4N electrons but not planar
But how do you know if it is a planar compound?
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
266
16.9 Heterocyclic aromatic compounds
If a cyclic compound with all p orbitals effective overlapping, plus with 4N+2 pi electrons, but a
carbon atom is replaced by a heteroatom (N, O, and S atoms). They are called heteroaromatic
compounds.
Pyridine
Pyridine is an aromatic nitrogen analogue of benzene. The nonbonding pair of electrons on nitrogen
replaces benzenes bond to a hydrogen atom. These nonbonding electrons are in an sp
2
hybride
orbital in the place of the ring. They are perpendicular to the pi system and do not overlap with it.
Because it has an available pair of nonbonding electrons, pyridine is basic. In an acidic solution,
pyridine protonates to give the pyridinium ion. The pyridinium ions is still aromatic because the
additional proton has no effect on the electrons of the aromatic sextet. It simply bonds to pyridines
nonbonding pair of electrons.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
267
Pyrrole
Pyrrole is an aromatic five-membered heterocycle. The pyrrole nitrogen atom is sp2 hybridized, and
its unhybridized p orbital overlaps with the p orbitals of the carbon atoms to form a continuous ring.
The lone pair on nitrogen occupies the p orbital, and these electrons take part in the pi bonding
system (belong to the aromaticity).
Pyrrole is a much weaker base than pyridine, because the nitrogen lone pair belongs to aromaticity.
Once the pyrrole is protonated, the nitrogen atom is bonded to four adiferent atoms, requiring sp3
hybridization. Hence, the protonated pyrrole is nonaromatic.
Pyrimidine
Pyrimidine is a six-membered heterocycle with two nitrogen atoms positioned in a 1,3-arrangement.
Each nitrogen atom has its lone pair of electrons in sp
2
hybride orbital in the plane of the aromatic
ring. The lone pairs of electrons are basic, like the lone pair of pyridine.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
268
Imidazole
Imidazole is an aromatic five-membered heterocycle with two hitrogen atoms. The lone pair of one
of the nitrogen is an sp2 orbital that is not involved in the aromatic system, therefore, it is basic. The
other one uses it third sp2 orbital to bond to hydrogen. Like the pyrrole. This imizazole N-H nitrogen
is not very basic because it belongs to aromaticity. Once imidazole is protonated, the two nitrogens
become chemically equivalent. Either nitrogen can lose a proton and return to an imidazole
molecule.
Purine
Purine has an imidazole fused to a pyrimidine ring. Purine has three basic nitrogen atoms and one
pyrrole-like nitrogen.
Pyrimidine and purine derivatives serve in DNA and RNA to specify the genetic code. Imidazole
derivatives enhance the catalytic activity of enzyme.
Furan
The nitrogen atom, replaced by oxygen atom in pyrrole, is furan. It is an aromatic five-membered
heterocycle. The oxygen is sp
2
hybridized, and one of the lone pairs occupies an sp
2
hybride orbital.
The other pair occupies the unhybridized p orbital.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
269
Thiophene
Thiophene is similar to furan, with a sulfur atom in place of the furan oxygen. The sulfur atom uses
an unhybridized 3p orbital to overlap with the 2p orbitals on the carbon atoms. However, the
resonance energy of thiophene (29 kcal/mol) is larger than the one of furan (16 kcal/mol).
16.10 Polynuclear aromatic hydrocarbons
Naphthalene
Fused aromatic compound, with containing two fused benzene rings. Naphthalene has less than
twice the resonance energy of benzene. (60 kcal/mol; 30 kcal/mol per aromatic ring) [36 kcal/mol in
benzene]
Anthracene and phenanthrene
As the number of fused aromatic rings increase, the resonance energy per ring continues to decrease
and the compounds become more reactive. Anthrance (84 kcal/mol; 28 kcal/mol per aromatic ring),
phenanthrene (91 kcal/mol; 30 kcal/mol per aromatic ring)
Because they are not as strongly stabilized as benzene, anthracene and phenanthrene can undergo
addition reactions to give two fully aromatic benzene rings, as shown below.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
270
Some of larger polynuclear aromatic hydrocarbons are carcinogenic. Some derivatives (epoxidation
of areneoxides) could be attacked by nucleophile sites of DNA (bind to DNA). The resulting DNA
derivatives cannot be properly transcribed. On replication, they cuase errors that produce mutation in
the gene. Some benign application is to serve as antitumor agents. Others malign effect is its DNA
mutation agents.
16.12 Fused heterocyclic compounds
The following fused heterocyclic aromatic rings are frequently found in nature. Some are skeleton of
amino acids, addictives, and drugs.
16.13 Nomenclature of benzene derivatives
Benzene derivatives have been isolated and used for over 100 years, such as salicylic acid. Salicylic
acid is the precursor of aspirin (lunched in 1898 by Bayer AG), which is the first drug on the market.
Therefore, historical common names are frequently used until now, and almostnever by systematic
IUPAC names.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
271
Dissubstituted benzene are named using the prefixes, ortho-, meta-, para- to specific the subatitution
patterns. Remember, when you type, it is ltalic.
Phenyl, benzyl, benzoyl, and aryl groups
16.15 Spectroscopy of aromatic compounds
IR
C=C stretch around 1600 cm
1
C=C-H stretching is 3030 cm
1
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
272
NMR
o 7 ~8
Mass
The most common mass spectral fragmentation of alkylbenzene derivatives is the cleavage of a
benzylic bond to give a resonance-stabilized benzylic cation. The base peak is at m/z 91, from the
benzyl cation, which may arrange to give the aromatic tropylium ion.
UV
The most characteristic part of the spectrum is the band centered at 254 nm, called the benzenoid
band.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
273
Chapter 17 Reactions of aromatic compounds
17.1 Electrophilic aromatic substitution
Like alkenes, electrophilic addition of alkenes gives addition product, where the alkenes serve as
the nucleophile (Nuc). Benzene, with 6 pi electrons, would also like to serve as the nucleophile to
attack the electronphile. However, benzene has resonance energy and is stable in an aromatic system.
With very strong electrophiles, benzene could serve as an\ nucleophile (Nuc) to undergoes
electrophilic aromatic substitution reaction through a resonance-stabilized carbocation, called sigma
complex.
Step 1:
Step 2:
PhH (benzene) as a Nuc to attack an electrophile. It results to form a sigma complex. To regain the
aromaticity, loss of a proton to gives the substitution product, where a proton (H
+
) was replaced by
an electronphile (E
+
). This type of reactions is called electrophilic aromatic substitution.
Brain-storming
Have you any ideas about the nucleophilic aromatic substitution? Benzene has 6 pi electrons
density (full of electrons), is it possible that benzene could serve as an electrophile? How?
We will see in section 17.12
The following reactions are classified as electrophilic aromatic substitution reactions.
Halogenation
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
274
Nitration
Sulfonation
Alkylation (Friedel-Craft alkylation)
Acylation (Friedel-Craft acylation)
17.2 Halogenation of benzene
General mechanism (bromination as example)
Recall
Electrophilic addition of alkenes
Electrophilic aromatic substitution with only bromine is not possible. Addition of bromine to
benzene is endothermic because it requires the loss of aromatic stability. Catalyst, such as FeBr
3
, acts
as a Lewis acid and converts bromine to a strong electrophile by forming a complex and polarizing
the BrBr bond.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
275
When the FeBr
3
was added as the Lewis acid
Bromination of benzene
Chlorination of benzene (AlCl
3
as Lewis acid)
Iodination of benzene (HNO
3
as oxidizing agent)
It requires an acidic oxidizing agent to oxidize I
2
to give iodine cation (I
+
).
17.3 Nitration of benzene
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
276
Preliminary step: formation of the nitronium ion, NO
2
+
Please draw the following steps by yourself. (similar to those in bromination)
Another application:
Nitration of benzene following by reduction is frequently used to make aniline, which is the best
method for adding an amino (NH
2
) group to an aromatic ring.
17.4 Sulfonation of benzene
Fuming sulfuric acid is the common name for a solution of 7% SO
3
in H
2
SO
4
. Sulfur trioxide is the
anhydride of sulfuric acid, meaning that the addition of water to SO
3
to gives H
2
SO
4
. Although it is
uncharged, sulfur trioxide is a strong electrophile. *Please draw the mechanism of sulfonation of
benzene by yourself.*
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
277
Sulfonation is economically important because alkylbenzene sulfonates are widely used as
detergents.
Desulfonation
Sulfonation is reversible, and a sulfonic acid group may be removed from an aromatic ring by
heating in dilute sulfuric acid. In practice, steam is often used as a source of both water and heat for
desulfonation.
Protonation of the aromatic ring: hydrogen (H) -deuterium (D) exchange
We could prove that a reaction has occurred by using a deuterium ion (D
+
) rather than a proton and
by showing that the product contains a deuterium atom in place of hydrogen. This experiment is
easily accomplished by adding SO
3
to some D
2
O (heavy water) to generate D
2
SO
4
.
The reaction is reversible and at equilibrium the final products reflect the D/H ratio of the solution.
This reaction serves as a synthesis of benzene-d
6
(C
6
D
6
), a common NMR solvent.
~Please jump to~
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
278
17.10 The Friedel-Crafts alkylation
Similar process could be applied to alkylation of benzene, in which a new carbon-carbon bond is
formed. It is the best way to make alkylbenzene.
The first step is the formation of a carbocation
*Please draw the following steps by yourself.*
Using others carbocation sources.
As you see the above equation, a carbocation could be generated from alkyl halides with Lewis acids.
Last semester, which reactions you have studied involving the generation of carbocation? Where?
Method 1 (electrophilic addition of alkene)
Alkenes are protonated by HF to give carbocations. Fluoride ion is a weak nucleophile and does not
immediately attack the carbocation. Once the benzene is present, electrophile aromatic substituent
occurs.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
279
Method 2 (dehydration of alcohols)
Alcohols are another source of carbons for Friedel-Crafts alkylation. Alcohols commonly from
carbocations when treated with Lewis acids, such as boron trifluoride (BF
3
). The BF
3
used in this
reaction is consumed and not regenerated. A full equivalent of the Lewis acid is needed, so we say
that the reaction is promoted by BF
3
rather than catalyzed by BF
3
.
Limitations of Friedel-Crafts alkylation
Limitation 1
It works only with benzene, activated benzene derivatives, and halobenzenes. Strongly
electron-withdrawing group (EWG, de-activating group) such as nitro (NO
2
), sulfonic acid
group (SO
3
H), carbonyl (C=O) on the benzene ring would pull the electron density away from the
benzene ring and result in deactivation of benzene. Eventually, electrophilic aromatic will NOT
occur.
Limitation 2
Once the carbocation is formed, it may undergo rearrangement. Be very very careful! As a result,
only certain alkylbenzenes can be made using the Friedel-Crafts alkylation. tert-Butylbenzene,
siopropylbenzene, and ethylbenzene could be made through this approach. However, consider what
happens, if we need to prepare n-propylbenzene using this synthetic approach?
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
280
n-Propyl chloride is used in the reaction, but isopropylbenzene is formed, which is not the desired
product.
Limitation 3
As you know that alkyl groups is an electron-donating group (EDG, activating group). Once the
first alkyl group is built, it will increase the electron-density of the benzene ring (alkylbenzene),
which is more reactive than starting material (benzene). Multiple alkylation reactions will occur to
give a mixture of di-alkylbenzene, trialkylbenzene. That is indeed a problem for a successful
reaction.
What can we do in this situation?
17.11 The Friedel-Crafts acylation
The Friedel-Carfts acylation is analogues to the Friedel-Carfts alkylation, except that the reagent is
an acyl chloride instead of an alkyl halide. In the present of aluminium chloride, the reaction of takes
place to give a phenyl ketone (acylbenzene).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
281
An acyl group means a carbonyl group with an alkyl attached.
The Fridel-Crafts acylation require acyl chloride for substitution. An acyl chloride could be prepared
by reactions of the corresponding carboxylic acids with thionyl chloride. Therefore, acyl chlorides
are also called acid chlorides.
The first step is formation of an acylium ion
One of the attractive features of the Fridel-Crafts acylation is the deactivation of the product toward
further substitution. The product (acylbenzene) has a carbonyl group (a deactivating group,
electron-withdrawing group) bond to the aromatic ring. The acylation stops after one substitution,
which is different from Fridel-Crafts alkylation. Friedel-Crafts acylation overcomes two of the three
limitations of the alkylation: The acylium ion is resonance-stabilized, so that no rearrangements
occur.
The Gatterman-Koch formylation: synthesis of benzaldehyde
A formyl group could not be made through Friedel-Crafts acylation, since the formyl chloride is
unstable. In situ generation of formyl chloride by carbon monoxide and HCl gas with a catalyst of
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
282
CuCl and Aluminum chloride could serve for the reagents, which is called Gatterman-Koch
formylation
Comparison of Friedel-Crafts alkylation and acylation
Alkylation Acylation
Will not occur when strongly deactivated
benzene
Will not occur when strongly deactivated
benzene
Involving carbocations, rearrangement may
occur
Not possible to rearrange,
Over-alkylation is a problem Not possible to react further
How do you synthesize alkylbenzenes that cannot be made by Friedel-Carfts alkylation? We use the
Friedel-Crafts acylation to make the acylbenzene, then we reduce the acylbenzene using following
two methods!
Two methods are frequently used for this transformation. One is an acidic condition (the
Clemmensen reduction). The other is a basic condition (the Wolff-Kishner reduction). They are
complementary methods.
The Clemmensen Reduction: Acidic condition
Treatment with aqueous HCl and amalgamated zinc (zinc treated with mercury salts)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
283
The reagents and conditions for the Clemmensen reduction are similar to those used to reduce a nitro
group to an amine.
Wolff-Kishner Reduction: Basic condition
(Section 18.20, Wade 8
th
, p. 864)
Ketone or aldehyde compounds that cannot survive treatment with hot acid can be deoxygenated
using Wolff-Kishner reduction. The ketone and aldehyde is converted to its hydrozone, followed by
treatment of a strong base such as KOH or KO
t
-Bu in high boiling point of solvent, such as ethylene
glycol around 140200
o
C to give the reduction product.
Mechanism will be discussed later when we reach chapter 18.
~Now back to ~
17.5 Nitration of toluene: the effect of alkyl substitutents
It is important to know the rate of substitution reaction starting from mono-substituent benzene.
Some substituents (activating group, such as CH
3
, OH, OCH
3
) speed up the reaction. Other
substituents (deactivating groups, such as Cl, NO
2
) retard the reaction. Toluene reacts about 25
times faster than benzene under the same conditions.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
284
Consider the following two nitration reactions with two different substrates.
When the starting material containing a methoxy (OMe) group, which is an activating group.
Ortho-, and para- disubstituted product are obtained predominately. When the starting material
containing a nitro (NO
2
) group, which is a deactivating group. Meta- disubstituted product is
obtained predominately.
17.6 Activating, ortho, para-directing substituents
Alkyl groups, alkoxyl groups, amino groups are activating groups, which would like to direct the
second substitution at ortho- and para- positions.
In the case of ortho or para attack, shift of an unshared electron pair from the oxygen to the positive
carbon allows the positive charge to be delocalized even further, onto the oxygen. (see the
structure in the dashed boxes) No such structures are possible for meta attack. Therefore, the
methoxy (-OCH
3
) group is ortho-, para-directing.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
285
[Ortho/para intermediates have four resonance-stabilized structures, which is far more stable
than meta structure (only three resonance-stabilized structures)]
Now, we can generalize this observation. All groups with unshared electrons on the atom attached
to the ring are ortho, para-directing. In each of them, the atom attached to the aromatic ring has an
unshared electron pair. Notice: even a halide atom is considered as a electron-withdrawing
group, it has an unshared electron pair, it is still a ortho-, para-directing group.
In alkyl group, the reaction can proceed via the most stable carbocation (3
o
) intermediate, all alkyl
groups are also ortho, para-directing. Please draw the mechanism by yourself. [Nitration of toluene]
17.7 Deactivating, meta-directing substituents
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
286
Nitro, sulfonic acid, cyano, ketone or aldehyde, ester, quaternary ammonium are de-activating
groups, which would like to direct the second substitution at meta- positions.
In the dashed boxes, there are two adjacent positive charges. The intermediate is highly unstable,
because the charges repel each other. No such intermediate is present for meta substitution. Meta
substitution is preferred.
[Ortho/para structures have an unstable resonance structure with two adjacent positive
charges]
A substitution on a benzene ring has its greatest effect on the carbon atoms prtho and para to the
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
287
substituent. An electron-donating substituent activates primarily the ortho and para positions, and an
electron-withdrawing substituents deactivates primarily the ortho and para positions.
17.8 Halogen substitutions: deactivating, but ortho, para-directing
There is one exception for this general rules. The halobenzene (with the halogens, F, Cl, Br, and I),
they are strongly electron withdrawing, therefore, the halogens are ring deactivating. But they have
unshared electrons pair, they are ortho, para directing group. Remember this.
17.9 Effect of multiple substituents on electrophilc aromatic substitution
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
288
What about if there are two substituents on the benzene, which groups (either activating, or
deactivating) dominate the electrophilic aromatic substitution?
Activating groups are usually stronger directors than deactivating groups, which are classed into
three groups, as following order (strong to weak)
1. Powerful ortho-, para-directing groups that stabilize the sigma complexes. OH, OR, NR
2
.
2. Moderate ortho-, para-directing groups, such as alkyl groups and halogens.
3. All meta directing groups.
Here are some examples:
Steric effect has to be considered, for example:
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
289
17.12 Nucleophilic aromatic substitution
We have learned electrophilic aromatic substitution, in which a benzene ring serves as the
nucleophile. However, in nucleophile aromatic substitution, a benzene ring (now as an electrophile)
has to be attacked by an electron pair, even though it has many pi electron density? Consider how a
benzene ring derivative could serve as an electrophile?
Nucleophiles can displace halide ions from aryl halides, particularly if there are strong
electron-withdrawing groups ortho- or para- to the halide.
There are two possible mechanisms. One is addition-elimination mechanism through sigma
complex. The other is elimination-addition mechanism through benzyne intermediate.
The addition-elimination mechanism
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
290
Step 1: attacked by a nucleophile to give a resonance-stabilized sigma complex
Step 2: Loss of the leaving group gives the product.
The mechanism showed how nitro groups ortho and para to the halogen help to stabilize the
intermediate. Without strong resonance-withdrawing groups in these positions, formation of the
negatively charged sigma complex is unlikely.
The benzyne mechanism: elimination-addition
Under extreme conditions, unactivated halobenzenes react with strong bases. For example: A
commercial synthesis of phenol (the Dow process) involvs treatment of chlorobenzene with
sodium hydroxide and a small amount of water in a pressurized reactor at 350
o
C.
Bromobenzene reacts with sodium amide (NaNH
2
, an extremely strong base) to give aniline,
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
291
Ph-NH
2
. This reaction does not require high temperature. However, A clue to the mechanism is
provided by the products are a 1:1 mixture of meta-, and para-toluidine. Two products can be
explained by an elimination-addition mechanism, called the benzyne mechanism.
Benzyne mechanism
Amide ion is a strong nucleophile, attacking at either end of the weak, reactive benzyne triple bond.
Subsequent protonation gives toluidine.
17.13 Aromatic substituents using organometallic reagents
In section 10.9, we discussed that organocuprate reagents (Gilman reagents) can couple with alkyl
halides. However, organocuprate reagents also couple with aryl and vinyl halide to make substituted
benzenes and elongated alkenes.
Coupling using organocuprate reagent
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
292
Here are some examples:
One of the feathers is that it involves sp
2
-sp
2
carbon bond, or sp
2
-sp
3
carbon bond coupling (from sp
2
carbon cuprate)
sp
2
-sp
2
carbon bond coupling
sp
2
-sp
3
carbon bond coupling
Furthermore, palladium-catalyzed coupling reactions, such as Heck reaction, Suzuki reaction,
Negishi reaction, are also important in organic synthesis. Professor R. F. Heck, Professor A, Suzuki,
and Professor E.-I. Negishi are 2010 Nobel Prize winners in Chemistry. You will learn in the class of
Advanced Organic Chemistry (AOC).
17.14 Addition reaction of benzene derivatives
Catalytic hydrogenation of aromatic rings
Catalytic hydrogenation of benzene to cyclohexane takes place at elevated temperature and pressures,
often catalyzed by ruthenium or rhodium-based catalysts. Di-substituted benzenes usually give
mixtures of cis and trans isomers.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
293
Birch reduction
Similar to the Na/liquid NH
3
reduction of alkynes to trans-alkenes, benzene derivatives are
reduced to non-conjugated cyclohexa-1,4-dienes by treatment with sodium (Na) or lithium (Li) in a
mixture of liquid ammonia and an alcohol.
Mechanism (single electron transfer): solvated electron
Step1: Addition of an electron, followed by a proton abstraction from alcohol.
Step 2: Another addition of single electron, , followed by a proton abstraction from alcohol
Electron-withdrawing substituents stabilize the carbanions, while electron-donating substituents
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
294
destabilize them. Therefore, Therefore, reduction takes place on the carbon atoms bearing
electron-withdrawing substituents, such as those containing carbonyl groups, not on the carbon
bearing electron-donating substituents, such as alkyl or alkoxyl groups.
Reduction on the carbon having EWG.
Reduction on the carbon without EDG
17.15 Side chain reaction of benzene derivatives
Benzylic position is one of the reactive positions in all benzene derivative.
Permanganate oxidation
This oxidation is occasionally useful for making benzoic acid derivatives, as long as any other
functional groups are resistant to oxidation. Functional groups such as NO
2
, halogens, -COOH,
and SO
3
H usually survive this brutal oxidation.
Side-chain halogenation
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
295
Alkylbenzenes undetgoes free-radical halogenation much more easily than alkanes because
abstraction of a hydrogen atom at a benzylic position gives a resonance-stabilized benzylic radical.
Chlorine radical is too reactive, even |-carbon may chlorinated.
The bromination radical is not reactive as a chlorine radical, therefore, it is more selective. It is a
common way to make a benzylic position with a bromide atom, such as benzyl bromide, source of a
lachrymator.
Nucleophilic substitution at the benzylic position
Allylic halides are more reactive than most alkyl halides in both S
N
1 and S
N
2 reactions. Benzylic
halides are also more reactive in these substitutions.
First-order reactions (S
N
1 reaction)
It require ionization of the halide to give a carbocation. In the case of a benzylic halide, the
carbocation is resonance-stabilized.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
296
A typical reaction of benzyl halide undergoes S
N
1 reaction.
Second-order reactions
During the S
N
2 displacement on a benzylic halide, the p orbital partially bonds with the
nucleophile and the leaving group also overlaps with the pi electrons of the ring. This stabilizing
conjugation lowers the energy of the transition state, increasing the reaction rate. Therefore, benzylic
halides are about 100 times more reactive than primary alkyl halides in S
N
2 displacement reactions.
17.16 Reactions of phenols
Much of the chemistry of phenols is like that of aliphatic alcohols. Esterification of salicylic acid
with acetic acid (or acetic anhydride) gave the acetylsalicylic acid (aspirin). Phenoxide ion could be
methylated with dimethyl sulfate.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
297
Oxidation of phenols to quinones
Phenols undergo oxidation, but they give different. Chromic acid oxidation of a phenol gives a
conjugated 1,4-diketone called a quinone.
Hydroquinone (benzene-1,4-diol) is easily oxidized to give 1,4-benzoquinone with silver bromide
(AgBr). His is the process of file developer (a hydroquinone solution).
Electrophilic aromatic substitution of phenols
Of course you must know, what are the products of the following electrophilic aromatic substitution?
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
298
Chapter 18 Ketones and aldehydes
Compounds containing carbonyl groups (C=O) are of central importance to organic chemistry,
biochemistry, and biology.
18.2 Structure of carbonyl group
The unhybridized p orbital overlaps with a p orbital of oxygen to from a pi bond The double bond
between carbon and oxygen is similar to an alkene C=C double bond, except that the carbonyl
double bond is short, stronger, and polarized.
The double bond of the carbonyl group has a larger dipole moment because oxygen is more
electronegative than carbon. This polarization of the carbonyl group contributes to the reactivity of
ketones and aldehydes: The positively carbon atom acts as an electrophile (Lewis acid), and the
negatively polarized oxygen acts as a nucleophile (Lewis base).
18.3 Nomenclature of ketones and aldehyses
IUPAC name ketone
Replace the final e in the alkane name with one. The alkane name becomes alkanone. In open
chain ketones, number the longest chain that includes the carbonyl carbon from the end closest to the
carbonyl group. In cyclic ketones, the carbonyl ketones, the carbonyl carbon atom is assigned the
number 1.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
299
IUPAC name aldehyde
Replace the final -e of the alkane with al. An aldehyde carbon is at the end of a chain, so it is
number 1. If the aldehyde group is a substituent of a large unit (usually a ring), the suffix
carbaldehyde is sued.
A ketone or aldehyde can also be named as a substituent on a molecule with a higher priority
functional group as its root. A ketone or aldehyde carbonyl is named by prefix oxo- if it is included
as part of the longest chain in the root name. When an aldehyde CHO group is a substituent and not
part of the logest chain. Is is named by the prefix formyl.
Common names:
Ketones common names are formed by naming the two alkyl groups to the carbonyl group.
18.4 Physical properities of ketones and aldehydes
Ketones and aldehydes have pairs of electrons and can act as hydrogen bond acceptors with other
compounds having O-H and N-H bonds.
Fromaldehyde is a gas at room temperature, so it is often stored and used as a 40% aqueous solution
called formalin. When dry formaldehyde is needed, it can be generated by heating one od its solid
derivatives, usually trioxane or paraformaldehyde. Trioxane is a cyclic trimer, containing three
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
300
formaldehyde units. Paraformaldehyde is a linear polymer. These solid derivatives are heated,
formaldehyde is spontaneously generated through heating.
18.5 Spectroscopy of ketones and aldehydes
IR
Ketones is 1710 cm
1
. (large dipole moment, strong peak)
Aldehydes is 1725 cm
1
, with a set of two low-frequency C-H stretching absorptions around 2710,
and 2810 cm
1
.
Conjugation lowers the carbonyl stretching frequencies of ketones and aldehydes because the
conjugation reduce the electron density of the carboyl pi bonds and is about 1685 cm
1
. Ring
strain raising the carbonyl stretching frequency in cyclic ketones. Because decreasing bond angle
increases the p character of C-C bond, s character of C=O bond is increasing. Therefore, C=O bond
becomes stronger, increasing C=O stretching frequency.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
301
1
H NMR
o-Hydrogen is around 2.1 ~2.4 ppm. Characteristic aldehyde peak is about 9 ~10 ppm.
13
C NMR
Characteristic peak of carbonyl of ketone and aldehyde is around 200 ppm. o carbon atom is about
3- ~40 ppm in
13
C NMR.
Mass spectra
A ketone and an aldehyde may lose an alkyl group to give a resonance-stabilized acylium ion
(through o-cleavage), ike the acylium ion that serves as the electrophile in the Fredel-Crafts
acylation.
McLafferty rearrangement
A common fragmentation with carbonyl compounds is generated from cleavage between the | and
carbons to give a resonance-stabilized carbocation.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
302
The base peak is at m/z 44, from loss of a fragment of mass 28, which is an even number. This
fragment is lost through a process called the McLafferty rearrangement, involving a cylic
intramolecular transfer of a hydrogen atom from the -carbon to the carbonyl oxygen.
The McLafferty rearrangement is a characteristic fragmentation of ketones and aldehydes as long
as they have hydrogens.
18.7 Review of syntheses of ketones and aldehydes
A: oxidation of alcohols
Others reagents are also used for making alcohol, such as Na
2
Cr
2
O
7
/H
2
SO
4
, PCC, PDC, Swern,
Dess-Martin periodinane (DMP).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
303
B: Ozonolysis of alkenes
C: Friedel-Carfts acylation
It is a method to make alkyl and aryl ketones.
Gatterman-Koch formylation
D: Hydration of alkynes
D-1: Catalyzed by acid and mercuric salts
Hydration of a terminal alkynes is a convenient way of making methyl ketones.
D-2: Hydroboration-oxidation of alkynes
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
304
18.818.10 Syntheses of ketones
~From carboxylic acids~
Organolithiums are so reactive toward carbonyls that they attack the lithium salts of carboxylate
anions to gives dianions. The first equivalent of organolithium deprotonated the acidic proton, then
second organolithium attacked the carbonyl to form the dianion. Protonation of the dianion forms the
hydrate of a ketone, which quickly loses water to give the ketone.
~From nitriles~
A Grignard or ortganolithium reagent attacks a nitrile to give the magnesium salt of an imine. Acidic
hydrolysis of the imine leads to the ketone. Note that the ketone is formed during the hydrolyxis
after any excess Grignard reagent has been destroyed. Thus the ketone is not attacked. [reverse of
acid-catalyzed imine formation]
Aluminum hydrides can reduce nitriles to the corresponding aldehydes. Diisobutylaluminum
hydride, abbreviated (i-Bu)
2
AlH or DIBAL-H is commonly used for the reduction of nitriles.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
305
R CN
1) (i-Bu)
2
AlH
R
H
N
Al(i-Bu)
2
H
3
O
+
nitrile
aluminumcomplex
R
H
O
+
NH
4
+
aldehyde
CN
1) (i-Bu)
2
AlH
2) H
3
O
+ H
O
~From acid chloride~
Aldehydes tend to be more reactive than acids. In normal cases, reducing agents are strong enough to
reduce acids also reduce aldehydes even faster.
Strong reducing agents like LiAlH
4
reduce acid chlorides all the way to primary alcohols. Lithium
tri-tert-butoxyaluminum [Li
+
Al (O-t-Bu)
3
H] hydride is a milder reducing agent that reacts faster
with acid chlorides than with aldehyse.
~From esters~
Diisobutylaluminum hydride (DIBAL-H) reduces esters directly to aldehydes at low temperature
(78
o
C). Cold DIBAL-H usually does not reduce the aldehyde further. The initial reaction froms an
aluminum complex that is stable toward further reduction, but hydrolyze to the aldehyde n the
aqueous workup.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
306
Synthesis of ketones
Grignard and organolithium reagents add to acid chlorides to give ketones, but they add again to the
ketones to give tertiary alcohols. How to stop at the ketone stage?
A weaker organometallic reagent is needed. One that reacts faster with acid chlorides than with
ketones. Lithium dialkylcuprates and others organocuprates are available choices.
18.11 Reactions of ketones and aldehydes:
The common reaction of ketones and aldehyde is nucleophilic addition, addition of a nucleophile
and a proton across the C=O double bond. The reactivity of the carbonyl group arises from the
electronegativity of the oxygen atom and the resulting polarization of the carbon-oxygen double
bond. The electrophile carbonyl carbon atom is sp2 hybridized and flat, leaving it relatively
unhindered and open to attack from either face of the double bond.
With strong nucleophiles, two examples of nucleophilic addition of ketone and aldehydes were
discussed before.
A: Grignard reagent attack the carbonyl to give an alkoxide, followed by subsequent protonation
to give an alcohol.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
307
B: Hydride ion (H
) act as the nucleophile to give an alkoxide, followed by protonation to from an
alcohol.
With weak nucleophiles, such as water and alcohols, can add to activated carbonyl groups under
acidic conditions. A carbonyl group is a weak base, and it can become protonated in an acidic
solution. A carbonyl group that is protonated is strongly electrophilic, inviting attack by a weak
nucleophile.
In most cases, aldehydes are more reactive than ketones toward nucleophile addition. The enhanced
reactivity of aldehydes is due to an electronic effect and a steric effect. Notice that an aldehyde has
only one electron-donated alkyl group, making the aldehyde carbonyl group slightly more
electron-poor abd electrophilic (the electronic effect). An aldehyde has only one bulky alkyl group,
leaving the carbonyl group more exposed toward nucleophilic attack. Especially with a bulky
nucleophile, the product of attack on the aldehyde is less hindered than the product from the ketones
(the steric effect).
Sodium triacetoxyborohydride [NaBH(OAc)
3
] is a mild reducing agent that reduces aldehyde
much more quickly than ketones. It can be used to reduce aldehyde in the presence of ketones.
Sodium cyanoborohydride (NaBH
3
CN) is another mild reducing agent. The reactivity of Sodium
cyanoborohydride (NaBH
3
CN) is less than NaBH
4
, but more than NaBH(OAc)
3
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
308
18.12 The Wittig reaction
In 1954, Professor Georg Wittig discovered a way of adding a phosphorus-stabilized carbanion to a
ketones and aldehyde. The intermediate undergoes elimination to an alkene.
Mechanism
Step 1: a phosphorus ylide formation. The phosphorus-stabilized carbanion is an ylide. An ylide is a
molecule that bears no overall charge but has a negatively charged carbon atom bonded to a
positively charged heteroatom.
Step 2: attack the carbonyl to give a betaine. A betaine is an unusual compound because it contains a
negatively charged oxygen and a positively charged phosphorus on adjacent carbon.
Step 3: formation of four-membered ring oxaphosphetane. Phosphorus and oxygen form strong
bonds and the attraction of opposite charges promotes the fast formation of oxaphosphetane.
Step 4: The oxaphosphetane collapses to give the alkene and triphenylphosphine oxide.
Triphenylphosphine oxide is exceptionally stable, and the conversion of triphenylphosphine to
triphenylphosphine oxide provides the driving force of this reaction.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
309
It is noticed that the Wittig reaction is a reverse reaction of ozonolysis (oxidative cleavage of
alkene to give aldehydes or ketones). Remember!!
Example 1:
O
Ph
3
P CH
2
CH
2
O
3
, thenMe
2
S
Wittig reaction
Ozonolysis
Example 2:
18.13 Hydration of ketones and aldehydes
In an aqueous solution, a ketone or an aldehyde is in equilibrium with its hydrate, a germinal diol.
With most ketones, the equilibrium favors the unhydrated keto form of the carbonyl.
Aldehydes are more likely than ketones to from stable hydrates. The electrophilic carbonyl group of
a ketone is stabilized by its two electron-donating alkyl groups, but an aldehyde carbonyl has only
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
310
one stabilizing alkyl group. Formaldehyde, with no electron-donating groups, is even less stable than
other aldehydes.
These stability effects are apparent in the equilibrium constants for hydration of ketones and
aldehydes. Ketones have values of K
eq
of about 10
4
to 10
2
. Formaldehyde is around 40.
Trichloroacetaldehyde is about 500.
18.14 Formation of cyanohydrins
Hydrogen cyanide (HCN) is a toxic, water-soluble with the pKa of 9.2Its conjugated base is the
cyanide ion, which is a strong base and a strong nucleophile. It attacks ketones and aldehyde to give
addition products called cyanohydrins. Cyanohydrins formation is reversible, the equilibrium may
or may not favor the cyanohydrin, dependent on the general reactivity of carbonyl.
formaldehyde >other aldehydes >ketones
One the most useful application of cyanohydrins is that they hydrolyze to give o-hydroxy acids
under the acidic conditions.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
311
18.15 Formation of imines
Under dehydration conditions, amine reacts with a ketone or an aldehyde to give an imine. Imines
are nitrogen analogues of ketones and aldehydes. Imines are involved as synthetic intermediates in
synthesis and biosynthesis. Like amines, imines are basic. A substituted imine is also called a Schiff
base.
Mechanism
Step 1: Acid catalyzed addition of the amine to the carbonyl group to give carbinolamine.
[Reacting with aldehyde, acid is not necessary for the imine formation, however, reacting with
ketones, acid is absolutely needed.]
Step 2: acid-catalyzed dehydration
It is noticed that the acid is used in the catalytic amount. If the solution is too acidic, however, the
amine becomes protonated and non-nucleophilic, which inhibits the first step.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
312
18.16 Condensations with hydroxylamine and hydrazines
Like imine formation, others amine derivatives could also react with ketones or aldehydes to give a
variety of nitrogen-containing compounds. Some of these compounds are the main skeleton of dyes.
Anhydrous hydrazine is a common rocket fuel.
18.17 Formation of acetals
With the same mechanism of imine formation, if the amine is replaced by an alcohol, what the
product will be obtained?
Step 1: Acid catalyzed addition of the alcohol to the carbonyl group. A hemiacetal is formed, like
the carbinolamine in imine formation
Step 2: acid-catalyzed dehydration to give a resonance-stabilized carbocation (we like to call it, an
oxycarbonium ion; a carbocation stabilized by adjacent oxygen)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
313
Step 3: this carbocation (the oxycarbonium ion) is attacked by the second alcohol to give acetal.
**Some key-points need to be emphasized again.
1. Have you found that the product is not a compound containing C=O double bonds? Why?
2. Since this reaction is reversible, if the product is treated with acidic aqueous solution, what
product will you expect to get?
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
314
3. If the acetal formation is carried out in the same molecule (intramolecular cyclization, not
intermolecular formation)?
4. If the alcohol is replaced with an diol, what is the product?
18.18 Using acetals as protecting groups
The acetal formation is carried out in acidic conditions. That means an acetal is labile (unstable) in
acidic solutions, however, it is very stable in basic conditions and stable to nulceophilic attack.
Why? The acetal is a sp
3
hybridization. No pi bond could be used to undergo nucleophilic addition.
Therefore, the acetals could be used to serve as the protecting groups of the carbonyl groups.
Please consider how to prepare the target compound, shown below! Obviously, it is NOT possible to
prepare a Grignard reagent with an aldehyde functionality, because both ketone and aldehyde groups
will be attacked by the Grignard reagent.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
315
[Remember: Grignard reagents are not compatible with any carbonyl groups]
Requirements of a protecting group:
1. Easy to put on
2. Compatible with the desired reaction conditions
3. Easy to take off
To keep the aldehyde group surviving in these conditions, the aldehyde group needs to be protected
during the Grignard reaction. The acetal is one of the most useful protecting groups of the carbonyls.
Protecting groups is very important in organic synthesis. An extremely useful book:
Greene's Protective Groups in Organic Synthesis, 4th Edition,
Editors: Peter G. M. Wuts, Theodora W. Greene, Wiley, 2006.
18.19 Oxidation of aldehydes
Common oxidants, such as bleach (sodium hypochlorite, NaOCl), cromic acid (H
2
CrO
4
),
permanganate (HMnO
4
), peroxy acid (H
2
O
2
). Because aldehydes oxidize so easily, mild reagents
such as Ag
2
O can oxidize them selectively in the presence of other oxidizable functional group.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
316
Silver ion, Ag
+
, oxidizes aldehydes selectively in a convenient functional group test for aldehydes.
Tollens test (Silver mirror reaction) is normally used to identify an unknown compound with an
aldehyde functionality.
18.20 Reduction of ketones and aldehydes
Hydride reduction
Sodium borohydride (NaBH
4
) is preferred for simple reduction of ketones and aldehydes. Lithium
aluminum hydride (LiAlH
4
) also accomplishes these reduction, but it is a more powerful reducing
agent. Sodium cyanoborohydride (NaBH
3
CN) and sodium triacetoxyborohydride
[NaHH(OAc)
3
] are less reactive than NaBH4, but it selectively reduces aldehyde or imine even in
the presence of ketones.
Catalytic hydrogenation
The most common catalyst is a finely divided hydrogen-bearing form of nickel made by treating a
nickel-aluminum alloy with a strong sodium hydroxide solution. Pt and Rh catalysts are also used
for hydrogenation of ketones and aldehydes.
Deoxygenation
A deoxygenation replaces the carbonyl oxygen atom of a ketones or aldehyde with two hydrogen
atoms, reducing the carbonyl group past the alcohol stage all the way to a methylene group.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
317
Deoxygenation cab be accomplished by either the Clemmensen reduction (under acidic conditions)
ro the Wolff-Kishner reduction (under basic condition)
Clemmensen reduction (acidic condition)
We have discussed that Friedel-Crafts alkylation sometimes have problems in rearrangement. To
avoid this problem. Cheimsts used Friedel-Crafts acylation +Clemmensen reduction to achieve
the same purpose. The actual reduction occurs by a complex mechanism on the surface of zinc.
Wolff-Kishner reduction (basic condition)
Compounds that cannot survive treatment with hot acid can be deoxygenated using the
Wolff-Kishner reduction. The ketone or aldehyde is first converted to its hydrazine, which is then
heated with a strong base such as KOH or potassium tert-butoxide in a high-boiling point solvent
Mechanism
Step 1: ketone with hydrazine to give hydrazone.
Step 2: proton transfer from nitrogen to carbon atom.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
318
Step 3: remove second proton from nitrogen atom to release nitrogen gas to from a carbanion, which
is quickly reprotonated to give the product.
Several others methods will be discussed in Advanced Organic Chemistry.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
319
Chapter 19 Amines
Amines are compounds containing nitrogen atoms. As a class, amines include some of the most
interesting biological compounds. Because of their high degree of biological activity, many amines
are used as drugs and medicines.
Alkaloids are an important group of biological active amines, either isolated fromnature or mostly
synthesized by plants to protect them from being eaten by insects and other animals. Some
representative alkaloids are shown below.
19.2 Nomenclature of amines
Amines are classified as primary (1
o
), secondary (2
o
), tertiary (3
o
), and quaternary ammonium
salts (4
o
).
Common names
Common names of amines are formed from the names of the alkyl groups bonded to nitrogen,
followed by the suffix amine.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
320
In naming amines with more complicated structures, the NH
2
group is called the amino group.
Aromatic and heterocyclic amines are generally known by historical names. Phenylamine is called
aniline.
Some nitrogen heterocycles are important in organic chemistry
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
321
19.3/4 Structure and physical properties of amines
Ammonia has a slightly distorted tetrahedral shape. A one pair of nonbonding electrons occupies one
of the tetrahedral positions. Bond angle of ammonia is 107
o
, and trimethylamine is 108
o
.
A tetrahedral amine with three different substituents (and a lone pair) is nonsuperimposable on its
mirror image, and appears to be a chirality center. In most cases, however, we cannot resolve such an
amine into two enantiomers because the enantiomers interconvert rapidly. This interconversion takes
place by nitrogen inversion, in which the one pair moves from one face of the molecule to the other.
Interconversion of (R)- and (S)-ethylmethylamine is shown below. In namine the enantiomers of
chiral amines, the Cahn-Ingold-Prelog Convention is used, with the nonbonding electron pair
having the lowest priority.
Although most simple amines cannot resolved into enantiomers, several types of chiral amines can
be resolved.
1. Amines whose chirality stem from the presence of asymmetric carbon atoms. (nitrogen is not the
chirality center)
2. Quaternary ammonium salt with asymmetric nitrogen atoms. (Inversion of configuration is not
possible because there is no lone pair to undergo nitrogen inversion)
3. Amines that cannot attain the sp
2
hybrid transition state for nitrogen inversion. (Not possible to
attain the 120
o
bond angles for nitrogen inversion)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
322
Amines are strongly polar because the large dipole moment of the lone pair of erlectrons adds to the
dipole moments of the CN and HN bonds. Primary and secondary amines have N-H bonds,
allowing them to from H-bonding. Pure tertiary amines cannot engage in hydrogen bonding because
they have no N-H bonds.
19.5/19.6 Basicity of amines/Effects on amine basicity
An amine could serve as a nucleophile and act as a base either. The following pK
a
of R
3
N
+
H is quite
important.
Amine pK
a
of
R
3
N
+
H
Classified
ammonia 9 normal amine
ethylamine 10 normal amine
pyrrole -1 nitrogen belongs to aromaticity
pyrrolidine 11 normal amine
piperidine 11 normal amine
pyridine 5 nitrogen lone pair NOT belongs to aromaticity
imidizole 7 nitrogen lone pair NOT belongs to aromaticity
How to quickly remember these values? Before that we need to know wat are K
b
and pK
b
.
How to affect the equilibrium?
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
323
1. Substitution by alkyl groups
Alkyl group are electron-donating groups, which donate electron density to the ammonium
ion(cation), and result in stabilization of the positive charge on the nitrogen. Hence, the equilibrium
partially favors the right side. K
b
increase, therefore, pK
b
decrease!
2. Resonance effect on basicity
Arylamines (anilines and their derivatives) are much weaker bases than simple aliphatic amines.
This reduced basicity is due to resonance delocalization of the nonbonding electron in the free
amines.
Resonance effects also influence the basicity of pyrrole. Pyrrole is a very weak base because it is
aromatic. When the pyrrole nitrogen is protonated, pyrrole loses its aromatic stabilization. Therefore,
protonation on nitrogen is unfacorable.
3. Hybridization effects
In section 9.6, we have learned that electrons are held more tightly by orbitals with more s character.
Unsaturated amines tend to be weaker bases than simple aliphatic amines. In pyridine, for
example, the nonbonding electrons occupy an sp2 orbital, with greater s character and more tightly
held electrons than those in the sp3 orbital of an aliphatic amine. Pyridines nonbonding electrons are
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
324
less available for bonding to a proton. Pyridine on protonation does not lose its aromaticity, while
pyrrole on protonation does lose its aromaticity, therefore, pyridine is stronger base than pyrrole.
The effect of increased s character on basicity is even more pronounced in nitriles with sp
hybridization. Acetonitrile is a very weak base.
19.7 Salts of amines
Simple amine salts are named as the substituted ammonium salts. Formation of amines salts can be
used to isolate and characterize amines. We can use the formation of amine salts to separate amines
from less basic compounds. This is what we called acid-base extraction.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
325
19.8 Spectroscopy of amines
We have learned before, however, we could memory..
The following partial IR spectra correspond to a primary amine, a secondary amine, and an alcohol.
Give the functional group for each spectrum.
Mass spectra
Stable compounds containing only carbon, hydrogen, oxygen, chlorine, bromine, and iodine give
molecular ions with even mass numbers. Most of their fragments have odd mass numbers.
Nitrogen has odd valence and an even mass number. In fact, whenever an odd number of nitrogen
atoms are present in a molecule, the molecular ion has an odd mass number. Most of the
fragments have even mass number.
19.9 Reactions of amines with ketones and aldehydes
Same in section 18.15
19.10 Aromatic substitution of arylamines and pyridine
Electrophilic aromatic substitution of arylamines
Amino groups are strong activating groups, therefore, the nonbonding electrons on nitrogem
stabilize the o complex when attack occurs at the ortho or para positions.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
326
An example of electrophilic aromatic substitution of aniline
Exception: When you wish to carry out the nitration of aniline,.
Strongly acidic reagents protonate the amino group, giving an ammonium salt that bears a full
positive charge. The NH
3
+
is strongly deactivating (and meta-allowing. Strongly acidic reagents are
unsuitable for substitution of anilines. Oxidizing acids (such as nitric and sulfuric acid) may oxidize
the amino group. Protection of amino groups is necessary.
[Supplementary materials]-organic synthesis
An intermediate in Woodwards synthesis of reserpine. Reserpine an indole alkaloid antipsychotic
and antihypertensive drug
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
327
Synthesis
While amino group is protected as an amide, which is still an electron-donating group. There is no
problem in regioselectivity of nitration.
Electrophilic aromatic substitution of pyridine (Py as nucleophile)
Pyridine resembles a strongly deactivated benzene in aromatic substitution reactions. Friedel-Craft
reactions fail completely, and other substitutions require unusually strong condition.
[Pyridine would form complex structurte with FeCl
3
and deactivate the substitution reaction.]
Deactivation results from the electron-withdrawing effect of the electronegative nitrogen atom.
Its nonbonding electrons are perpendicular to the t system, and they cannot stabilize the positively
charged intermediate. When pyridine does react, it gives substitution at the 3-position, analugues to
the meta substitution shown by deactivated benzene derivatives.
Attack at 3-position (positive charge spread over three carbon and not on the nitrogen)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
328
Attack at 2-position (not favored, unstable intermediate with only six electrnos on nitrogen)
Nucleophilic aromatic substitution of pyridine (Py as electrophile)
If there is a good leaving group at either the 2-position or the 4-position, a nulceophile can attack
and displace the leaving group. The intermediate is stabilized by delocalization of the negative
charge onto the electronegative nitrogen atom. This stabilization is not possible if attack occurs at
the 3-position.
Nucleophilic attack at the 2-position (or the 4-position) forms a stabilized intermediate.
Nucleophilic attack at the 3-position (negative charge not on the nitrogen)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
329
19.11/19.12/19.13 Alkylation/Acylation/Sulfonation of amines
Alkylation
The disadvantage of direct alkylation lies in stopping it at the desired stage. Some amine molecules
will react once. Eventually, A complex mixtures result. Exhaustive alkylation (over alkylation)
gives a tetraalkylammonium salt. To obtain the monoalkylated product, a large excess of ammonia is
needed.
Acylation
The amide produced in this reaction usually does not undergoe further acylation. Because amides are
stabilized by a resonance structure that involvs nitrogens nonbonding electrons and place a positive
charge on nitrogen. As a result amides are much less basic and less nucleophilic than amines. Acyl
groups is sometimes used as protecting group for amines.
Sulfonation
Like acyl chloride, sulfonyl chlorides are the acid chlorides of sulfonic acids. Amides of sulfonic
acids are called sulfonamides. The Sulfa drugs are a class of sulfonamides used as antibacterial
agents. Sulfanilamide is synthesized from acetamide and foundf to be effective against infection.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
330
19.14 Amines as leaving groups: The Hofmann elimination
An amino group can be converted to a good leaving group by exhaustive methylation. Upon
treatment of a strong base. To provide the base, the quaternary ammonium iodide is converted to the
hydroxide salt by treatment with silver oxide.
Heating of the quaternary ammonium hydroxide results in E2 elimination and formation of an alkene.
This elimination of a quaternary ammonium hydroxide is called the Hofmann elimination. In the
Hofmann elimination, the product is commonly the least-substituted alkene.
Recall: (Chapter 7)
Zaitsevs rule: The most substituted product predominates, because the most substituted alkene is
usually the most stable.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
331
The Hofmann eliminations preference for the least-substituted alkene stems from several reasons.
One of the most important is the elimination of a bulky leaving group in E2 reaction mechanism.
First: how many |-hydrogens it has
C
3
-H2 / C
1
-H3, total 5 |-hydrogen
To abstract the C
3
-H is NOT preferred because the required E2 anti-coplanar conformation is NOT
stabled. However, to abstract C
1
-H is preferred because the required E2 anti-coplanar conformation
is absolutely stabled.
19.15 Oxidation of amines: The Cope elimination
Amines are notoriously easy to oxidize, and oxidation is often a side reaction in amine synthesis.
Amines also oxidize during storage in contact with the air. Preventing air oxidation is one of the
reasons for converting amines to their salts for storage or use as medicines. The following partial
structures show some of the bonding and oxidation states of amines
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
332
The mechanisms of amine oxidation are not well characterized, partly because many reaction paths
(especially those involving free radicals) are available. Secondary amines are easily oxidized to
hydroxylamines. Tertiary amines are oxidized to amine oxides.
Because of the positive charge on nitrogen, the amine oxide may undergo a Cope elimination, much
like the Hofmann elimination of a quaternary ammonium salt. The amine oxide acts as its own base
through a cyclic transition state, so a strong base is not needed. The Cope elimination generally
gives the same orientation as Hofmann elimination, resulting in the least-substituted alkene.
19.16/19.17 Reactions of amines with nitrous acid to form diazonium salts
Some amines compounds could serve as medicines. It could also be converted to a variety of useful
molecules. Amines could react with nitrous acid to form diazonium salts. Thess diazonium salts
could be used to provide different functional groups, even dyes.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
333
Nitrous acid is unstable, it is generated in situ by mixing sodium nitrite (NaNO
2
) with cold and
dilute HCl.
Formation of nitrosonium ion (
+
N=O).
Reaction with primary amines with nitrous acid, via nitrosonium ion, to give diazonium salts.
Step 1
Step 2
Step 3
N-nitrosoamines have been shown to cause cancer in laboratory animals. These finding have
generated concern about the common practice of using sodium nitrile (NaNO
2
) to preserve meats
such as bacon, ham, and hot dogs. When the meat is eaten, sodium nitrile combines with stomach
acid tof from nitrous acid, whi9ch can convert amines in the food to N-nitroaimes.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
334
19.17 Reaction of arenediazonium salts
Arenediazonium salts are relatively stable in aqueous solution around 0-10
o
C. The diazonium group
can be replaced by many functional groups. Arenediazonium salts are formed by diazotizing a
primary aromatic amine. Primary aromatic amines are commonly prepared by nitrating (NO
2
) an
aromatic ring, then reducing the nitro group to an amino group (NH
2
).
Phenol
The hydroxyl group of water replace N
2
, forming a phenol. This is a useful laboratory synthesis of
phenols (ArOH) because (unlike nucleophilic aromatic substitution) it does not require strong
electron-withdrawing substituents or powerful bases and nucleophiles.
Sandmeyer reaction
Copper (I) salts have a special affinity for diazonium salts. CuCl, CuBr and CuCN react with
arenediazonium salts to give aryl chlorides (ArCl), aryl bromides (ArBr), and aryl cyanides (ArCN).
The use of copper salts to replace arenediazonium groups is called the Sandmeyer reaction. The
case of ArCN is also an excellent method for attaching another carbn substituent to an aromatic ring.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
335
Deamination of anilines
Hypophosphrous acid (H3PO2) reacts with arenediazonium salts, replacing the diazonium group
wioth a hydrogen. In effect, it can be regarded as de-amination or aromatic amine.
Problem:
How you could convert toluene to 3,5-dibromotoluene in good yield.
Direct bromination is not possible!
Correct answer:
Introducing a strong activating group (amino NH
2
) at the para position is the choice, and then
following by the de-amination reaction. But how to introduce an amino group? Nitration then
reduction.
Diazo coupling
Arenediazonium ions act as weak electrophiles in electrophilic aromatic substitutions. The
compound with N=N linkage are called azo compounds. Because they are weak electrophiles,
diazonium salts react only with strongly activated rings (such as derivatives of aniline and phenol)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
336
Two substituted aromatic rings into conjugation with an azo group, which is a strong chromophere.
Most azo compounds are strongly colored, and they make excellent dyes, known as azo dyes. The
following reaction is the synthesis of methyl orange (a common indicator in chemical laboratory)
The sulfonate (SO
3
) or carboxylate (COO
) promote the solubility in water.
19.18/19.19/19.20 Synthesis of amines
Six different methods are included in this section
1. Reductive amination
2. Acylation then reduction
3. Gabriel amine synthesis (for primary amines)
4. Azide reduction (for primary amines)
5. Nitrile reduction (for primary amines)
6. Nitro reduction
1. Reductive amination
Reductive amination is the most general amine synthesis, capable of adding a primary or secondary
alkyl group to an amine. It is a two steps procedure.
Step 1: amines react with aldehydes or ketones to give imines
Step 2: reduce the imine to amine.
Primary amines
Primary amines result from condensation of hydroxylamine with a ketone or an aldehyde, followed
by reduction of the oxime. Hydroxylamine is used in place of ammonia because most oximes are
stable, easily isolated compounds, The oxime is reduced using catalytic reduction, lithium aluminum
hydride, or zinc and HCl.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
337
Secondary amines
Condensation of a primary amine with a ketone and aldehyde froms an N-substituted imine (a
Schiff base). Reduction of the imine, using either LiAlH
4
or NaBH
4
gives a secondary amine.
Tertiary amines
Condensation of a secondary amine with a ketone and aldehyde gives an iminium salt. Because
iminium salts are frequently unstable, so they are rarely isolated. In situ reduction of iminium salts
in the reaction solution is necessary. To reduce iminium salts rather than reduce the carbonyl group
of ketones or aldehydes, a reducing reagent that must be less reactive to reduce ONLY iminium
salts in the presence of carbonyl groups. Two reagents are frequently used in this transformation.
There are sodium triacetoxyborohyride [NaBH(OAc)
3
] and sodium cyanoborohydride
[NaBH
3
CN], both are much less reactive than sodium borohydride [NaBH
4
].
2. Acylation then reduction
Thesecond general synthesis of amines is acylation then reduction. Acylation of the starting amines
by an acid chloride gives an amide, which is much less nucleophilic and unlikely to over-acylate.
Reduction of the amide by lithium aluminum hydride (LiAlH
4
) gives the corresponding amine.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
338
Acylation-reduction converts ammonia to a primary amine, convert a primary amine to a secondary
amine, or a secondary amine to a tertiary amine. The added alkyl group is always primary alkyl
group (1
o
) because the carbon bonded to nitrogen is derived from the carbonyl group of the amide,
reduced to a methylene (-CH
2
-) group.
An example:
3. Gabriel amine synthesis (for primary amine)
Primary amines are frequently used for starting materials for synthesis of secondary and tertiary
amines. Several methods are introduced here.
Direct alkylation
The S
N
2 reaction of amines with alkyl halides is complicated by a tendency for over-alkylation to
from a mixture of monoalkylated and polyalkylated products, as we discussed in section 19.11.
Simple primary amines can be synthesized, however, by adding a halide or tosylate (must be a good
S
N
2 substrate) to a large excess of ammonia.
Siegmund Gabriel (University of Berlin) developed a useful method to prepare primary with
over-alkylation. Phthalimide has one acidic N-H proton (pKa =8.3) that is abstracted by potassium
hydroxide to give the phalimide ion, which is a protected form of ammonia that cannot alkylate
more than once. Alkylation of this phthalimide ion gives the N-alkyl phthalimide. Generation of the
primary amine is finished by treatment of hydrazine (H
2
N-NH
2
). The side product is a stable
hydrazide of phthalimide.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
339
4. Azide reduction (for primary amines)
Two other sources of nitrogen atom are azide ion (
N
3
) and cyanide ion (
CN). However, the azide
ion introduces (after reduction) an NH
2
group, and the cyanide ion introduce a CH
2
-NH
2
group
with one more carbon.
Azide ion (
N
3
) is an excellent nucleophile that displace leaving groups from unhindered primary
and secondary alkyl halides and tosylates. The product are alkyl azide (RN
3
), which have no
tendency to react further. Azides are easily reduced to primary amines, either by LiAlH
4
or by
catalytic hydrogenation (Pd/ H
2
or Pt/H
2
).
Notice that sodium azide (NaN
3
) or alkyl azides can be EXPOLSIVE (commonly used for airbag
of automobiles), so they are directly used for the next step without purification.
Azide ion also reacts with a variety of electrophiles. The following is the example to make
|-hydroxyl amine (aminoalcohol) from the reaction of the epoxide with sodium azide.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
340
5. Nitrile reduction (for primary amines)
Like azides, cyanide ion (
CN) is also a good nucleophile to react with alkyl halides and tosylate.
The product is called nitrile (R-CN), with one more carbon. The nitriles are reduced to primant by
LiAlH
4
or catalytic hydrogenation.
An example:
We have learned that the ketones or aldehydes could react with cyanide to generate cyanohydrin.
Upon reduction, the cyanohydrin could be converted to |-hydroxyl amine (aminoalcohol).
6. Nitro reduction (normally used to make aromatic amine)
Normally, aromatic amines, such as aniline, are easily made from nitration of an aromatic ring,
followed by reduction of the nitro group to amino group. Aliphatic amines are normally made
from the methods discussed above.
The most common reason for reducing aromatic nitro compounds is to make substituted anilines.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
341
Here is an example to make benzocaine (a topical anesthetic)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
342
Chapter 20 Carboxylic Acids
The combination of a carbonyl group and a hydroxyl on the same carbon atom is called a carboxyl
group. Compounds containing the carboxyl group are distinctly acidic and are called carboxylic
acids, which could serve as the hydrogen bond donor and hydrogen bond acceptor.
20.2 Nomenclature of carboxylic acids
In common names, the position of substituents are named using Greek letters. Notice that the
lettering begins with the carbon atom next to the carboxyl carbon, the o carbon. With common
names, the prefix iso-is sometimes used for acids ending in the CH(CH
3
)
2
grouping.
In IUPAC nomenclature, the final e in the alkane name is replaced by the suffix oic acid. The
chain is numbered, starting with the carboxyl carbon atom, to give positions of substituents along the
chain. In naming, the carboxyl group takes priority over any of the other functional groups we
have discussed.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
343
Unsaturated acids are named using the name of the corresponding alkene, with the final e replaced
by oic acid. The stereochemical terms cis and trans (Z and E) are used as they are with other
alkenes. Cycloalkanes with COOH substituents are generally named as cycloalkanecarboxylic
acids.
Aromatic acids for the form Ar-COOH are named as derivatives of benzoic acid, Ph-COOH. As
with other aromatic compounds, the prefixes ortho-, meta-, and para- may be used give the
positions of additional substituents.
A dicarboxylic acid (also called a diacid) is a compound with two carboxyl groups. A common
mnemonic for these names is Oh! my such good apple pie, standing for oxalic, malonic, succinic,
glutaric, adipic, and pimelic acids.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
344
Benzenoid compounds with two carboxyl groups are named phthalic acids. Phthalic acid itself is the
ortho isomer. The meta isomer is called isophthalic acid, and the para isomer is called terephthalic
acid.
20.3 Structure and physical properities of carboxylic acids
The most stable conformation of formic acid is shown next. The entire molecule is approximately
planar. The O-H bond also lies in this plane, eclipsed with the C=O bond. Because one of the
unshared electron pairs on the hydroxyl oxygen atom is delocalized into the electrophilic pi system
of the carbonyl group. The delocalization forms are represented below.
Carboxylic acids boil at considerably higher temperatures than do alcohols, ketones, aldehydes of
similar weights. The high boiling points of carboxylic acids result from formation of a stable,
hydrogen-bonded dimer. This dimer contains an eight-membered ring joined by two hydrogen
bonds, shown below.
20.4 acidity of carboxylic acids
A carboxylic acid dissociates in water to giv e a proton and a carboxylate ion. The equilibrium
constant Ka for this reaction is called the acid-dissociation constant. Normally, the pKa of an acid
is about ~5 (acetic acid is 4.7), which is about 10
11
times more acidic than the most acidic alcohol.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
345
With the equations shown above, what would you expect that an acid is attached an
electron-withdrawing group (EWG)? Enhance or decrease the acid strength? And Why?
The electron-withdrawing group stabilize the carboxylate ion, therefore, the equilibrium favor the
right side and increase the acidity.
A nitro group (a electron-withdrawing group) increases the acidity, while a methoxy
substituted (a electron donating group) decrease the acidity. The nitro group has a larger effect
in the ortho and para position than in the meta position. Considering the nitrobenzene, which
positions are lees reactive toward electrophilic aromatic substitution reaction? A nitro group is
meta-directing group, isnt it !
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
346
20.5 Salts of carboxylic acids
A strong base can completely deprotonate a carboxylic acid. The product are a carboxylate ion, the
cation remaining from the base. The combination of a carboxylate ion and a cation is a salt of a
carboxylate acid.
Soap is a common example of carboxylate salts, consisting the soluble dodium salts of long-chain
fatty acids. Carboxylate salts of most other metal ions are insoluble in water. For example, when
soap is used in hard water containg calcium, magbesium, or ion ions, the insoluble carboxylate salts
precipitate out as hard-water scum.
Some purification methods take advantage of the different solubilities of acids and their salts.
Nonacidic impurities can be removed from a carboxylic acid using acid-base extractions, similar
with the method used in purification of amine/ammonium salts
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
347
20.6 Commercial sources of carboxylic acids
The most important commercial aliphatic acid is acetic acid. Vinegar is a 5% aqueous solution of
acetic acid. Vinegar for food is produced by fermentation of sugars and starches.
Industrial synthesis of acetic acid is relied on catalytic process. Ethylene is converted to
acetaldehyde under the condition of Wacker process, followed by another catalytic oxidation to
produce acetic acid. You will learn these processes in the class of Inorganic Chemistry.
Benzoic acid is used an ingredient in medication, a preservative in foods, and a starting material for
synthesis. Benzoic acid can be produced by the oxidation of toluene (PhCH
3
) with potassium
permanganate (KMnO
4
), nitric acid (HNO
3
), or other strong oxidants.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
348
Two important commercial diacid are adipic acid (hexanedioic acid) and tere-phthalic acid
(benzene-1,4-dicarboxylic acid). Adipic acid is used in making nylon 66, and terephathalic acid
is used to make polyesters. The industrial synthesis of these two acids uses benzene as the starting
material. Benzene is hydrogenated to cyclohexane, whose oxidation gives adipic acid. Terephthalic
acid is produced by the direct oxidation of para-yxlene in acetic acid using a cobalt-molybdenum
catalyst.
20.7 Spectroscopy of carboxylic acids
IR
In a saturated acid, this vibration occurs around 1710 cm
1
. In conjugated acids, the carbonyl
stretching frequency is lowered to about 1690 cm
1
. The O-H band of acids is around 3000 cm
1
,
which is different from the O-H band of alcohols in 3300 cm
1
.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
349
NMR
Carboxylic acids protons are the most deshielded protons we have encountered, absorption between
o 10 and 13. The protons on the o carbon atom absorb between o 2.0 and 2.5. The carbonyl carbon
atom absorbs around o 170 to 180, and the o carbon atom absorbs around o 30 to 40.
UV (not often to use for structure determination)
Saturated carboxylic acids have a weak n t* transition that absorbs around 200 to 215 nm.
Conjugated acids show much stronger absorptions. One C=C double bond conjugated with the
carbonyl group results in a spectrum with
max
around 200 nm, but with the molar absorptivity of
about 10,000. A second conjugated dou8ble bonds rasies the value of
max
to about 250 nm.
Mass spectrometry
The molecular ion peak of a carboxylic acid is usually small because favorable modes of
fragmentation are available. The most common fragmentation is loss of a molecule of an alkene
[molecular weight =28] (the McLafferty rearrangement). The ion that results from Malafferty
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
350
rearrangement has an even-numbered mass, as opposed to the odd-numbered ions that result
from loss of fragments.
20.8 Synthesis of carboxylic acids
1. Oxidation of alcohol and aldehydes
2. Oxidative cleavage of alkenes and alkynes
3. Side-chain oxidation of alkylbenzenes
4. Carboxylation of Grignard reagents
5. Hydrolysis of nitrile
6. Haloform of ketones (chapter 22)
7. Malonic ester synthesis of acids (chapter 22)
1. Oxidation of alcohols and aldehydes
Oxidized by chromic acid (H
2
CrO
4
, NaOCl)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
351
2. Oxidative cleavage of alkenes and alkynes
Cold, diluted potassium permanganate reacts with alkenes to give glycols. Warm,
concentrated permanganate solutions oxidize the glycols further, cleaving central
carbon-carbon bond.
With alkynes, either ozonolysis or a vigorous permanganate oxidation cleavages the triple bond to
give carboxylic acids.
3. Side-chain oxidation of alkylbenzenes
Side chains of alkylbenzenes are oxidized to benzoic acid derivatives by treatment with hot
potassium permanganate or hot chromic acid. Because this oxidation requires sever condition, it
is useful only for making benzoic acid derivatives with no oxidizable functional groups.
Oxidation-resistant functional groups such as Cl, NO
2
, SO
3
H, and COOH may be present.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
352
4. Carboxylation of Grignard reagents
Grignard reagents add to carbon dioxide to from magnesium salts of carboxylic acids. Addition
of dilute acid protonates these magnesium salts to give carboxylic acids. This method is useful
because it converts a halide (R-X) functional group to a carboxylic acid (R-COOH) functional
group with an additional carbon atom.
5. Hydrolysis of nitriles
Another method to convert an alkyl halide (R-X) (or tosylate, R-OTs) to a carboxylic acid
(R-COOH) with an additional carbon atom is to displace the halide with sodium cyanide
(NaCN) to give a nitrile (R-CN). Acid or basic hydrolysis of the nitriles gives a carboxylic acid.
This method is limited to halides and tosylates that are good S
N
2 electrophiles: usually primary
and unhindered.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
353
6. The haloform reaction (Chapter 22)
Methyl ketone is treated with a base to givea trihalo-ketone. Then, nucleophilic attack to the
carbonyl group gives the acid with the haloform as a side product.
7. Malonic ester synthesis (Chapter 22)
Substitution of malonic ester [CH
2
(CO
2
Et)
2
] gives o-alkyl malonic ester [R-CH(CO
2
Et)
2
].
Upon treatment with base (hydrolysis) and heat (release of carbon dioxide), the alkyl halide
(R-X) could be converted to a carboxylic acid (R-CH
2
-COOH) with two additional carbon
atoms.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
354
20.9 Reactions of Carboxylic acids and derivatives; nucleophilic acyl substitution
Ketones and aldehydes commonly react by nucleophilic addition to the carbonyl group; but
carboxylic acids (and their derivatives) more commonly react by nucleophilic acyl substitution,
where one nucleophilic replaces another on the acyl (C=O) carbon atom.
Acid derivatives differ in the nature of the nucleophile bonded to the acyl carbon: OH in the acid,
Cl in the acid chloride, OOCR in anhydride, OR in the ester, and NH
2
in the amide.
Nucleophilic acyl substitution is the most common method for intervonverting these derivatives. We
will learn in Chapter 21.
20.10 Condensation of acids with alcohols: The Fischer esterification
The Fischer esterification converts carboxylic acids and alcohols directly to esters by an
acid-catalyzed nucleophilic acyl substitution. The net reaction is replacement of the acid OH
group by the OR group of the alcohol.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
355
20.11 Esterification using diazomethane
Carboxylic acids are converted to their methyl esters very simply by adding an ether solution of
diazomethane. The only by-product is nitrogen gas, and any excess diazomethane also evaporates.
Purification of the ester usually involves only evaporation of the solvent. Yields are nearly
quantitative in most cases.
However, diazomethane is a toxic, explosive yellow gas that dissolves in ether and is fairly safe to
use in ether solution. The mechanism is shown below.
Because diazomethane is hazardous in large quantities, it is rarely used industrially or in
large-scale laboratory reactions. The yields of methyl esters are excellent, however, so
diazomethane is often used for small-scale esterification of valuable and delicate carboxylic acids.
20.12 Condensation of acids with amines: Direct synthesis of amides
Amides can be synthesized directly from carboxylic acids, using heat to drive off water and force the
reaction to completion. The direct synthesis is an important industrial process, abd it often works
well in the laboratory.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
356
20.13 Reduction of carboxylic acids
1. LAH
2. Borane
Lithium aluminium hydride (LiAlH4 or LAH) are strong enough to reduce carboxylic acids
directly to primary alcohols. The aldehyde is an intermediate in this reduction, but it cannot be
isolated because it is reduced more easily than the original acid.
Mechanism:
Step 1: remove the acidic proton
Step 2: reduce the carbonyl
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
357
Step 3: To form the intermediate aldehyde, and then reduce the carbonyl again to from the alkoxide.
Two hydrogen atoms were belonged to the hydride sources.
Step 4: Protonation of the alkoxide to give the primary alcohol.
Borane also reduces carboxylic acids to primary alcohols. Borane (complex with THF) reacts with
the carbonyl group faster than with any other carbonyl function. It often gives excellent
selectivity, as shown by the following example, where a carboxylic acid is reduced while a ketone is
unaffected. (LiAlH
4
would also reduce the ketone.)
Reduction of carboxylic acids to aldehyde is difficult because aldehydes are more reactive than
carboxylic acids toward most reducing agents. A two steps process could be applied.
LiAlH(O-t-Bu)
3
is a weaker reducing agent than LiAlH
4
. It reduces acid chlorides to aldehydes
because acid chlorides are strongly activated toward nucleophilic addition of a hydride ion. Under
these conditions, the aldehyde reduces more slowly and can be isolated.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
358
20.14 Alkylation of carboxylic acids to form ketones
Carboxylic acids reacted with two equivalents of an organolithium reagent to give ketones. The first
equivalent of organolithium deprotonates the acid. The second equivalent adds to the carbonyl o give
a stable dianion. Hydrolysis of the dianion (by adding water) gives the hydrate of a ketone.
20.15 Synthesis and use of acid chlorides.
Halide ions are excellent leaving groups for nucleophilic acyl substitution. Therefore, acyl halides
are useful intermediates for making acid derivatives. Both the carbonyl oxygen and the chlorides
atom withdraw electron density from the acyl carbon atom, making it strongly electrophilie. Acid
chlorides react with a wide range of nucleophiles, generally through the addition-elimination
mechanism of nuelcophilic acyl substitution.
The best reagents for converting carboxylic acids to acid chlorides are thionyl chloride (SOCl
2
) and
oxalyl chloride [(COCl)
2
] because they from gaseous by-products that do not contaminate the
product. Oxalyl chloride is particularly easy to use because it boils at 62
o
C and any excess
evaporated from the reaction mixture.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
359
Mechanism:
Step 1: Either oxygen atom of the acid attack sulfur, replacing chloride by a mechanism that looks
like sulfurs version of nucleophilic acyl substitution. The product is an interesting, reactive
chlorosulfite mix-anhydride.
Step 2: This mix-anhydride undergoes nucleophilic acyl substitution by chloride ion to give the acid
chloride.
Esters from acyl chlorides
Acyl chloride reacts with alcohol to give esters. Pyridine (Py), triethylamine (TEA), or other bases
are often added to neutralize the HCl generated. Otherwise, alcohols (especially tertiary alcohols)
may dehydrate under strongly acidic conditions.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
360
Amides from acyl chlorides
Ammonia and amines react with acid chlorides to give amides through the same addition-elimination
mechanism. A base such as pyridine or NaOH is often added to prevent HCl from protonating the
amine. Protonation of the amine would result in the formation of ammonium chloride. The amine
loses its nucleophilicity.
[Supplementary material]
R OR
O
R Cl
O
R O
O
R
O
R NH
2
O
R OH
O
acyl
halide
acid
anhydride
ester amide acid
reactivity
(addition)
stability
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
361
Chapter 21 Carboxylic Acid Derivatives
Carboxylic acid derivatives are compounds with functional groups that can be converted to
carboxylic acids by a simple acidic or basic hydrolysis. Nitriles are also considered as derivatives,
because they have similar oxidation state of carbon, as to the carbon of carboxylic acid.
21.2 Structure and nomenclature of acid derivatives
Esters
The names of esters consist of two words that reflect their composite structure. The first word is
derived from the alkyl group of alcohol, and the second word from the carboxylate group of the
carboxylic acid.
Lactones
Cyclic esters are called lactones. A lactone is formed from an open chain hydroxyl acid in which the
hydroxyl group has reacted with the acid group to form an ester.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
362
The common names of lactones, used more often than IUPAC names, are formed by changing
theic acid ending of the hydroxyl acid to olactone. A Greek letter designates that carbon atom that
bears the hydroxyl group to close the ring.
Amides
The simple amide structure shows a nonbonding pair of electrons on the nitrogen atom. Amides are
only weakly basic. This lack of basicity can be explained by picturing the amide as a resonance
hybride of the conventional structure and a structure with a double bond between carbon and
nitrogen. For example: formamide gas a planar structure like an alkene. The C-N bond has partial
double bond character.
The from R-CO-NH
2
is called primary. The form R-CO-NHR is called a secondary amide or
N-substituted amide. The from R-CO-NR
2
is called tertary amide or N,N-disubstituted amides.
Drop the ic acid or oic acid suffix of the corresponding acid, and add the suffix amide. For
secondary and tertiary amides, treat the alkyl groups on nitrogen as substituents, and specify their
position by the prefix N-.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
363
For acids that are named as alkanecarboxylic acids, the amides are named by using the
suffix carboxamide.
Lactams
Like lactone, cyclicamides are called lactams. Lactams are named by adding lactam at the end of
the IUPAC name of the parent acid. Common names of lactams are formed by changing the ic acid
ending of the amino acid to olactam.
Nitriles
Nitrile contain the cyano group, CN. They are classified as acid derivatives because they hydrolyze
to give carboxylic acids and can be synthesized by dehydration of amides.
Although a nitrile has a lone pair of electrons o nitrogen, it is NOT very basic. A typical nitrile has a
pKb of about 24, requiring a concentrated solution of mineral acid to protonate the nitrile. We
explain this lack of basicity by noting that the nitriles lone pair resides in an sp hybride orbital, with
50% s character. This orbital is close to the nucleus, and these electrons are ttightly bound and
relatively unreactive.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
364
Common name of itriles are named by replacing the suffix ic acid with the suffix onitirle. The
IUPAC name is constructed from the alkane name with the suffix nitrile added.
Interconversion between acid and nitriles
We will learn these later!
Acid halides
Acid halides, also called acyl halides, are activated derivatives used for the synthesis of other
acid derivatives. The halogen atom of an acyl halide inductively withdraws electron density from the
carbonyl carbon, enhancing its electrophilic nature and making acyl halides particularly reactive
toward nucleopilic acyl substitution. The halide ion laso serve as a good leaving group.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
365
Acid anhydrides
The word anhydride means without water. An acid anhydride contains two molecules of an acid,
with loss of a molecule of water.
Although anhydrides are not reactive as acid halides. In an anhydride, the carboxylate group serves
these functions, such as chloride in acyl halides.
Anhydride nomenclature is very simple. The word acid is changed to anhydride in both the common
name and the IUPAC (rarely used).
Anhydride composed of two different acids are called mixed anhydride and are named by using the
names of the individual acids.
Nomenclature of multifunctional compounds
Following the priority
acid >ester >amide >nitrile >aldehyde >ketone >alcohol >amine >alkene,alkyne
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
366
Here are some examples:
21.3 Physical properties of carboxylic acid derivatives
Boiling point and melting points
In carboxylic acids are strongly hydrogen bonded in the liquid phase. The stable hydrogen-bonded
dimer has large effect on the boiling point. Nitrile has higher boiling points because it has a strong
dipolar association between adjacent cyano groups.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
367
Amides have surprisingly high boiling points and melting points compared with other compounds of
similar molecular weight, even higher than acid. The resonance picture shows a partial negative
charge on oxygen and a partial positive charge on nitrogen.
Solubility
Acid chloride and acid anhydride cannot be used in nucleophilic solvents such as water and alcohols.
However, they react with these solvents. Esters (EtOAc), tertiary amides (DMF), and nitriles
(CH
3
CN) are frequently used as solovents for organic reactions, because they provide a polar
reaction medium without O-H or N-H groups that can donate protons or act as nucleophiles.
21.4 Spectroscopy of carboxylic acid derivatives
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
368
Ester
Ester carbonyl groups absorb at about 1735 cm
1
. Conjugated ester absorb around 1710 to 1725 cm
1
and might easily be confused with simple ketones (1710 cm
1
) and aldehyde (1725 cm
1
), The
presence of both a strong carbonyl absorption is this region and a conjugated C=C absorption around
1620 to 1640 cm
1
suggests a conjugated ester.
Amides
Simple amides have much lower stretching frequencies, absorbing around 1640 to 1680 cm
1
. The
C=O bond of the amide carbonyl is somewhat less than a full double bond. Primary and secondary
amides have N-H bonds that give infrared stretching absorption in the region 3200 to 3500 cm
1
.
These absorption fall in the same region as the board O-H absorption of an alcohol, but the amide
N-H absorption are usually sharper.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
369
Lactones and Lactams
Unstrained lactones (cyclic esters) and lactams (cyclic amides) absorb at typical frequencies for
esters and amides. Ring strain raises the carbonyl absorption frequency. Cyclic ketones with
five-membered or smaller rings show a similar increase in carbonyl stretching frequency.
Nitriles
Nitriles usually absorb at frequencies slightly higher than 2200 cm
1
(to the left of 2200 cm
1
), while
alkynes usually absorb at frequencies slightly lower than 2200 cm
1
; and nitrile absorptions are
usually stronger because the CN triple bond is more polar than the alkyne CC triple bond.
Acid halides and anhydrides
Acids chlorides absorb at a high frequency, around 1800 cm
1
. Anhydride give two carbonyl
stretching 1800 and 1750 cm
1
. (CCC handout shows it absorbs at 1810 and 1760 cm
1
)
NMR spectroscopy
The following are typical absorption of acid derivatives in the proton NMR spectrum.
The spectrum of DMF (N,N-dimethylformamide) is shown below. It is noticed that the two methyl
groups are stereochemically non-equivalent. The two methyl groups appear as two singlets (not a
spin-spin splitting doublet). The two singlets result from hindered rotation about the amide bond.
The cisoid and transoid methyl groups interconvert slowly with respect to the NMR time scale.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
370
21.5 Interconversion of acid derivatives by nucleophilic acyl substitution
Through the addition-elimination process, the nucleophilic reagent adds to the carbonyl group to
produce a tetrahedral intermediate, which expels the leaving group to regenerate the carbonyl
group.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
371
Acid chloride are the most reactive acid derivatives, so they are easily converted to any of the other
acid derivatives. Using the scheme, it helps you to convert highly reactive acid derivative to highly
stable acid derivatives.
R OR
O
R Cl
O
R O
O
R
O
R NHR
O
R OH
O
acyl
halide
acid
anhydride
ester
amide
carboxylic acid
reactivity
(addition)
stability
high
low
low
high
leaving group / conjugated acid
Cl
O R
O
OR
NHR
OH
HCl
HO R
O
HOR
NH
2
R
H
2
O
Exercise:
1. From acid chlorides to anhydrides
2. From acid chloride to amides
3. From anhydrides to ester
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
372
4. From ester to amide
5. From amide to acid
21.6 Transesterification (from an ester to another ester)
R OR
O
R OR'
O
21.7 Hydrolysis of acid halides, anhydride, esters, amides
Hydrolysis of nitriles
Basic hydrolysis
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
373
Acid hydrolysis
Try to draw both mechanisms by yourself!!
2.8 Reduction of acid derivatives
Lithium aluminum hydride (LAH) reduces acids, acid chlorides, anhydrides, and ester to primary
alcohol. Acid chloride is so reactive that NaBH
4
is strong enough to reduce it to the primary
alcohol.
R OR
O
R Cl
O
R O
O
R
O
R NHR
O
R OH
O
acyl
halide
acid
anhydride
ester
amide
carboxylic acid
LiAlH
4
R OH
R OH
R OH
R OH
R NHR
NaBH
4
is also effective
in this case
exception, amine is
obtained
R H
O
Through the addition-elimination mechanism, the reactive intermediate is an aldehyde. The
aldehyde is more reactive than acid derivatives, it would be further reduced to an alkoxide. After
the reduction is complete, dilute acid is added to protonate the alkoxide to give the primary alcohol.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
374
How to obtain aldehyde from acid derivatives
Using LAH, it is not possible to obtain the aldehyde (to stop the mechanism at the aldehyde stage).
To successfully obtain the aldehyde, several methods were developed.
1. Aldehyde from acid chloride using LiAlH(O-t-Bu)
3.
Longer reaction time would reduce to
alcohols too.
2. Aldehyde from ester using (i-Bu)
2
AlH at low temperature (78
o
C). Need to quench at low
temperature! (semi-reduction)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
375
3. Aldehyde from nitrile using (i-Bu)
2
AlH at low temperature (78
o
C). Need to quench at low
temperature! (semi-reduction)
4. Reduce to alcohol, then oxidize the alcohol to the aldehyde.
Exception: reduction of amides to amines
Strong reducing agents (LAH) reduces amides and nitriles to amines, providing some of the best
synthetic routes to amines. Primary amides and nitriles are reduced to primary amines. Secondary
amides are reduced to secondary amines, and tertiary amides are reduced to tertiary amines.
The mechanism of this reduction begins like a typical nucleophilic acyl substitution, with hydride
adding to the carbonyl group to form a tetrahedral intermediatre. The nitrogen atom is a poor
leaving group, however, the former carbonyl oxygen atom, complexed with aluminum, is a fair
leaving group. The oxygen atom leaves with aluminum, giving an imine or iminium salt quickly
reduce to the amine.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
376
Nitriles are reduced to primary amines
Nitriles could be reduced to primary using hydrogenation (H
2
/Pt) or LAH. When nitriles is
treated with DIBAL-H at low temperature, and also quench at low temperature, it could be
reduced to imine (we call semi-reduction), which could be converted to aldehyde in acidic aqueous
solution.
21.9 Reactions of acid derivatives with organometallic reagents
Grignard and organolithium reagents add TWICE to acid chloride and esters to give alkoxides.
Protonation of the alkoxides gives tertiary or secondary alcohols.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
377
Thefirst equivalent gives the ketone, the second equivalent gives the alkoxides. Unless the original
ester is a formate (R =H), which gives a secondary alcohol. In each case, two of the groups on the
product are the same, derived from the organometallic reagents. (Key: once the carbonyl is formed,
the Grignard or organometallic reagent will attack the carbonyl)
How to get the ketone?
The reactions shown above are not possible to obtain the ketone (the intermediate). Any carbonyl
group is formed, the nucleophilic reagent will attack the carbonyl group to give the tertiary
alcohol after protonation. The ketone is far more reactive than acid derivatives toward nulceophilic
addition. To successfully obtain the ketone, several methods are developed to give the ketone (stop at
the stage of ketone).
1. From acyl chloride with dialkylcuprates (Gilman reagents). Gilman reagent is less reactive
than Grignard reagent. Gilman reagent does react with aldehydes, rarely react with ketones.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
378
1. From nitrile. Grignard reagent attacks the electrophilic cyano group to form the salt of an imine.
Acidic hydrolysis of the salt (in a separate step) gives the imine, which if further hydrolyzed to a
ketone.
21.10 Summary of acid chlorides
How to prepare acid chlorides? Thionyl chloride (SOCl
2
) and oxalyl chloride (COCl)
2
are the
most convenient reagents because they produce only gaseous side products.
Reaction of acid chlorides
One common feather of these reactions is the generation of hydrochloride. A base is sometimes used
to the sponge to remove the acid (HCl) and to drive the reaction to the right side.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
379
Acid chlorides react with strong reagents to give DOUBLE addition products. Relatively weak
reagents give the addition-elimination product.
Friedel-Crafts acylation
21.10 Summary of anhydride
Acetic anhydride
Acetic anhydride is produced at the rate of about 4 billion pounds per year, primary for the synthesis
of plastics, fibers and drugs (such as aspirin). The industrial synthesis begins by dehydrating acetic
acid to give ketene. Triethyl phosphate is added as a catalyst. Ketene (a gas at rt.) is fed directly into
acetic acid, where is reacts quickly and quantitatively to give acetic anhydride.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
380
General anhydride synthesis
To prepare mix anhydride, another method needs to be developed. The most general method for
making anhydride is the reaction of an acid chloride with a carboxylic acid or a carboxylate salt.
Anhydride undergoes many od the same reactions as acid chlorides.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
381
Some cyclic anhydrides are made simply by heating the corresponding diacid. A dehydrating agent,
such as acetyl chloride or acetic anhydride, is occasionally added to accelerate the reaction.
Some cyclic anhydrides are quite useful in synthesis. Cyclic anhydrides are used to make
difunctional compounds.
It can also provide additional functionality on the side chain of the aromatic product after
Friedel-Crafts acylation.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
382
Regarding the atom economic, one of the two acid molecules (one side of the anhydride) would loss
(as a leaving group). If a precious acid needs to be activated, is will waste your materials. Several
methods could be applied to overcome this problem.
Use of acetic formic anhydride
Formyl chloride (the acid chloride of formic acid) cannot be used for formylation because it quickly
decomposes to CO and HCl. Acetic formic anhydride is made from sodium formate and acetyl
chloride. The nucleophile would like to react primarily at the formyl group. Lacking a bulky,
electro-donating alkyl group, the formyl group is both less hindered and more electrophilic than the
acetyl group.
Use of pivaloyl acid (or called: 2-trimethylacetic acid)
The carbonyl of the pivaloyl group is too hindered to be attacked. Therefore, the nucleophile would
like to attack the other side of carbonyl group to give the desire product.
When the RCOOH is a precious acid.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
383
21.12 Summary of esters
Esters are among the most common acid derivatives. Warfarin is used as an anticoagulant.
Warfarin slows clotting of the blood, which can prevent heart attacks and strokes.
The order of ripe bananas comes from isoamyl acetate. Oil of wintergreen contains methyl
salicylate. Lavendor oil contains small amounts of coumarin, which gives depth and longevity of
their orders. Sperm whales use spermaceti to regulate their buoyancy and as a resonating chamber
for communicating underwater.
Synthesis of esters
Esters could be made from any of others acid derivatives. Methyl ester could also be made by
treatment of diazomethane.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
384
Reactions of esters
Formation of lactones
Simple lactones containing five- and six-membered rings are often more stable than the open-chain
hydroxyl acids. Such lactones form spontaneously under acidic conditions.
Macrolactones that are not energetically favored may be synthesized by driving the equilibrium
toward the products. A diluted benzene solution containing a trace amount of p-toluenesulfonic acid
(PTSA, TsOH) is used for the following reaction to remove water and shift the equilibrium to the
right.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
385
Polyester
Several things made from polyesters could be found in your life supplies. You clothes are made from
Dacron. Your DVD is made of Mylar. The electronic, such as cell phone, are made by Glyptal
polyester resin. The plastic bottle of your soft drink was made from PET.
21.13 Summary of amides
Synthesis of amides
Amides are the least reactive acid derivatives, and they can be made from any of the others. The
industrial synthesis of amide involves heating. However, amides are commonly made by the reaction
of acid chlorides and amines.
Reactions of amides
Amides are the most stable acid derivatives. From a synthetic point of view, their most important
reaction is the reduction to amines.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
386
Strong dehydrating agents can move the element of water from a primary to give a nitrile, which
provide one of the most common methods for the synthesis of nitriles. Phosphorus pentoxide (P
2
O
5
)
is the traditional reagent for this dehydration, but phosphorus oxychloride (POCl
3
) sometimes gives
better yields, which is common used in the laboratory.
Mechanism: Try to draw the reaction mechanism by yourself.
Lactams
Five-membered (-lactams) and six-membered lactams (o-lactams) often form upon heating or
adding a dehydrating agent to the appropriate -amino acids and o-amino acids. Lactams containing
smaller or larger rings do not form readily under these conditions.
|-lactams are usually reactive amides and are capable of acylating a variety of nucleophiles. The
considerable strain in the four-membered ring appears to be the driving force behind the usual
reactivity of |-lactams. The |-lactam ring is found in three important classes of antibiotics, all
isolated from fungi.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
387
The organic chemistry of drug action
Polyamides: Nylon 6,6 is made by mixing adipic acid and hexane-1,6-diamine.
21.14 Summary of nitriles
Synthesis of nitrile
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
388
Reactions of nitriles
21.15 Thioesters
A thioester is formed from a carboxylic acid and a thiol.
Thioesters are more reactive toward nucleophilic acyl substitution than normal ester, but less
reactive than acid chlorides and anhydrides.
The enhanced reactivity of thioesters results from two reasons. The first is resonance stabilization.
The second resonance form involves overlap between a 2p orbital on carbon and a 3p on sulfur.
The overlap is weak and relatively ineffective, leaving the C-S bond of a thioester weaker than the
C-O bond of an ester.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
389
The second is that an alkyl sulfide anion (RS
) is a better leaving group than an alkoxide anion
(RO
) because the sulfide is less basic than an alkoxide (or thiol is more acidic than alcohol). The
larger sulfur atom carries the negative charge spread over a larger volume of space. Hence, sulfur is
also more polarizable than oxygen, allowing more bonding as the alkyl sulfide anion is leaving.
Thioester are not prone to hydrolysis, yet they are excellent selective acylating agents, which
could be found in living systems. Many biochemical acylating involve transfer of acyl groups from
thioesters of coenzyme A (CoA). In fact, acetyl CoA serves as a water-stable equivalent of acetyl
chloride.
21.16 Carbonic acid/carbamic acid
Carbonic acid is formed reversibly whenever carbon dioxide dissolves in water. All carbonated
beverages contain carbonic acid in equilibrium with CO
2
and water.
Carbonate esters are diesters of carbonic acids
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
390
Polycarbonates
Lexan polycarbonate is a strong, clear polymer used tomake bulletproof winder and carsh helmets.
The diol used to make Lexan is a phenol called bisphenol A.
Ureas are diamides of carbonic acid. Carbamate esters (urethanes) are the stable ester of the
unstable carbamic acid, the monoamide of carbonic acid.
Many of carbamate esters are made from phosgene, acid chloride of carbonic acid. This method may
not causes the formation of both carbonates and carbamates.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
391
Another reliable method is to treat an alcohol or with an isocyanate, which is an anhydride of a
carbamic acid.
Several carbamates are frequently used for the protection of amino group (N-protecting group) in
amino acid (AA) synthesis, such as Cbz, Boc, or Fmoc. They all have the structure of carbamate.
Cbz is carbobenzoxy; Boc is tert-butyloxycarbonyl; Fmoc (fluorenylmethyloxycarbonyl)
R COOH
NH
2
amino acid (AA)
R COOH
HN O
O
Cbz (Z)-AA
R COOH
HN O
O
R COOH
HN O
O
Boc-AA Fmoc-AA
H
2
/Pd TFA Piperidine
abbreviation
remove
(deprotecting method)
Polyurethane are the material of latex-free gloves, which cause fewer allergic reactions.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
392
Chapter 22 (this is the last chapter in this course)
Condensations and alpha substitutions of carbonyl compounds
Carbonyl group is one of the most important functional group in Organic Chemistry. Two types of
reactions: substitution at the carbon atom next to the carbonyl group (called alpha substitution) and
carbonyl condensation.
Alpha substitution
Carbonyl condensation
22.2 Enols and enolate ions
Keto-enol tautomerism (enolization)
A proton on the o carbon atom is abstracted by a strong base to form a resonance-stabilized enolate
ion with the negative charge spread over a carbon atom and an oxygen atom. [Reprotonation can
occur either on the o carbon (returning to the keto form) or on the oxygen atom], giving a vinyl
alcohol, the enol form. Either base or acid could catalyze the keto-enol tautomerism.
Base-catalyzed enolization
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
393
Acid-catalyzed enolization
These two isomers, keto and enol isomers, occurring by the migration of a proton and the movement
of a double bond, is called tautomerism, and the isomers that interconvert are called tautomers.
Tautomers are true isomers (different compounds) with their atoms arranged differently.
In addition to its mechanistic importance, keto-enol tautomersim affects the stereochemistry of
o-carbon of the carbonyl compounds. A hydrogen atom on an o carbon may be lost and regained
through keto-enol tautomerism; such a hydrogen is said to be enolizable. A trace of acid and base
allows that carbon to inverts its configuration (racemization).
Formation and stability of enolate ions
Carbonyl groups dramatically increase the acidity of the protons on the o-carbon atom because
deprotonation gives a resonance-stabilized enolate ion. Most of the enolate ions negative charge
resides on the electronegative oxygen atom. The pKa for removal of an o proton from a typical
ketone or aldehyde is about 20. Still a ketone or aldehyde is less acidic than water (pKa =15.7) or an
alcohol (pKa =16 to 18).
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
394
In some cases, this eqilbrium mixture of enolate and base wont work, usually because the
base )hydroxid or alkoxide) reacts with the electrophile (E
+
) faster than the enolate does. To
overcome, we need a base to completely remove the o proton and convert the carbonyl compound to
its enolate before adding the electrophile. The most effective and useful base for this purpose is
lithium diisopropylamide (LDA), which is made by reaction of n-butyllithium and diisopropylamine.
LDA is about as basic as sodium amide (NaNH
2
), but much less nucleophilic because it is hindered
by the two bulky isopropyl groups. Therefore, it could only serve as a strong base not a strong
nucleophile. After deprotonation, the resulting lithium enolate salts are quite useful in synthesis.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
395
22.3 Alkylation of enolate ions
We have seen that carbon nucleophile, such as Grignard reagent) could attack tosylates by the SN
2
mechanism. However, an enolate has two nucleophilic sites (the oxygen and the o-carbon), it can
react at either of these sites.
LDA is a very bulky base and thus a poor nucleophile, so it generally does not react with the alkyl
halide or tosylate. Direct alkylation of enolate gives the best yields when only one kind of o
hydrogen can be replaced by an alkyl group. Mixtures of products may obtain if two different kinds
of o protons could be deprotonated. Aldehydes are not suitable for direct alkylation because they
undergo side reactions, such as Aldol reaction. (aldehydes are reactive enough toward nucleophilic
attack)
If there are more than one o hydrogens could be removed by LDA, over alkylation may occur. How
to solve the problem.
22.4 Formation and alkylation of enamines
A milder alternative to direct alkylation of enolate ions is the formation and alkylation of an enamine
derivative. An enamine ( a vinyl amine) is the nitrogen analogue of an enol. An enamine results from
the reaction of a ketone or aldehyde with a secondary amine.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
396
Enamines are intermediate in reactivity: more reactive than an enol, but less reactive than an enolate.
Enamines displace halides from reactive alkyl halides, giving alkylated iminium salts. The iminium
ions are reactive toward further alkylation or acylation. The alkylated iminium salt hydrolyzes to the
alkylated ketone.
The overall reactions are step-wise procedures of the o-alkylation of the carbonyl groups. Because
of the formation of iminium ion, it avoids over-alkylation.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
397
The enamine alkylation procedure is sometimes called the Stork reaction, after its inventor, Gilbert
Stork of Columbia University. The Stork reaction can alkylate or acylate the o position of a ketone,
using a variety of reactive alkyl and acyl halides.
The following sequence shows the acylation of an enamine to synthesize a |-diketone. Synthesis of
|-diketone could not prepared easily by the direct acylation of a ketone. Because the pKa of a
|-diketone is about 9 ~11, which is more acidic than the starting ketone (~20).
22.5 Alpha halogenations of ketones
The o-Alkylation reaction provides a method to make the carbon-carbon bond at the o-carbon.
However, the carbon-heteroatom (C-X) bond is not easy to make through similar mechanisms,
because of the polarity is reversed. The following methods provide a practical approach to make C-X
bond.
Base-promoted o-halogenation
When a ketone is treated with a halogen and base, an o-halogenation reaction occurs.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
398
The mechanism is shown below. Step 1: deprotonate the o proton to form an enolate
Step 2: base-promoted bromination
Through the mechanism, the product is the o-halo ketone, which is even more acidic than the
starting ketone. In base conditions, the o-halo ketone could be continuously deprotonated to from an
enolate, which is stabilized by the halogen (X). Actually, multiple halogenations do occur to give
further halogenations.
The halofrom reaction
With most ketones, base-promoted halogenations continue until the o carbon atom is completely
halogenated. Methyl ketones undergo halogenations three times to give trihalometjylketones.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
399
The trihalomethyl group can serve as a reluctant leaving group. The trihalomethyl ketone reacts with
hydroxide ion to give a tetrahedral intermediate that expels the trihalomethyl anion (-CX
3
), leaving a
carboxylic acid. A fast proton exchange gives a carboxylate ion and a haloform. (chloroform, CHCl
3
;
bromoform, CHBr
3
; or iodofrom CHI
3
).
The mechanism of haloform reaction
The problem is still reminded. It is not easy to make a C-X bond at o-carbon in basic conditions. So,
how to make mono-halogenated ketone? The answer is the acidic conditions.
Acid-catalyzed o-halogenation
One of the most effective procedures is to dissolve the ketone in acetic acid, which serves as both the
solvent and the acid catalyst. Acidic halogenation can selectively replace just one hydrogen or more
than one, depending on the amount of the halogen added. The mechanism is shown below.
We can stop the acid-catalyzed reaction at the mono-halo product because the halogen-substituted
enol intermediate is less stable than the unsaturated enol. Halogen has strong electronegativity,
therefore, it deactivated the halogen-substituted enol intermediate. Can you draw that?
An example is shown below.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
400
Question: Acid-catalyzed haogenation is synthetically useful for converting ketones to
o,|-unsaturated ketones, which are useful in Michael reactions Proposer a method for converting
cyclohexanone to cyclohex-2-en-1-one, an important synthetic starting material.
Answer:
22.6 o-Bromination of acids: The HVZ reaction
The HVZ reaction replaces a hydrogen atom with a bromine atom on the o carbon of a carboxylic
acid. The carboxylic acid is treated with bromine (Br
2
) and phosphorus tribromide (PBr
3
), followed
by water to hydrolyze the intermediate o-bromo acyl bromide. The mechanism is shown below.
Step1: the acid is converted to acyl bromide. The mechanism of this step is not shown in the
textbook. So, can you draw by yourself?
Step 2: The enol form of the acyl bromide reacts with bromide to give the o-bromo acyl bromide
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
401
This o-bromo acyl bromide serves as an activated intermediate (similar to an acid chloride) for the
synthesis of an o-bromo ester, o-bromo amide, or other derivative. If the o-bromo acid is needed,
a water hydrolysis completes the synthesis.
22.7 The Aldol reaction (Aldol condensation) of ketones and aldehyde
Under acidic basic conditions, the nucleophilic addition of an enolate to another carbonyl group
provides a product, |-hydroxyl ketone or aldehyde, (at this stage) is called an Aldol reaction.
Because it contains both an aldehyde group and the hydroxyl group of an alcohol. The aldol product
may dehydrate to an o,|-unsaturated carbonyl compound. The whole process is called Aldol
condensation. Condensation means a reaction combine two or more molecules, often with the loss
of a small molecules such as water or an alcohol.
Base-catalyzed aldol condensation
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
402
Acid-catalyzed aldol reaction
The aldol condensation is reversible, establishing an equilibrium between reactants and products.
22.8 Dehydration of aldol products (Aldol condensation)
Heating a basic or acidic mixture of an aldol product leads to dehydration of the alcohol functional
group. The product is a conjugated o,|-unsaturated aldehyde or ketone. An aldol condensation,
followed by dehydration, forms a new carbon-carbon double bond. Before the Wittig reaction was
discovered, the aldol condensation was probably the best method for join two molecules with a
double bond formation. Dehydration is usually exothermic because it leads to a conjugated system.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
403
Please draw the mechanisms of dehydration under basic or acidic conditions by yourself!
22.9 Crossed aldol condensations
When the enolate of one aldehyde (or ketone) adds to the carbonyl group of a different aldehyde or
ketone, the result is called a crossed aldol conbdensation. The situation is similar with the
dehydration of alcohols to form ether. (unsymmetric ether formation). Depending on the reaction
conditions, various proportions of the four possible products result.
With appropriated design, a crossed aldol condensation can be effectively if it is planned so that only
one of the reactants can form an enolate ion. Two successful crossed aldol condensation reactions are
shown below. One of the aldehydes has no o proton.
Example 1:
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
404
Example 2:
22.10 Aldol cyclization (Intramolecular aldol condensation)
Intramolecular aldol condensation are usually used to prepare cyclic unsaturated ketone.
Cyclopentenone
Cyclohexenone
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
405
Another example shows how the carbonyl group of the product may be outside the ring in some
cases.
Complete the following synthesis
22.11 Planning syntheses using aldol condensation
The following retrosynthetic analyses show you how the aldol products could be made from the
starting materials.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
406
22.12 The Claisen condensation (Claisen ester condensation)
The ester version of Aldol condensation is called Claisen condensation. Similar to aldol reactions,
the enolate of an ester would attack the carbonyl group of another ester molecule and resulted in
formation of a |-keto ester. Although no double bond (alkene) is formed, it does condense a
molecule of an alcohol. The overall reaction combines two ester molecules to give a |-keto ester.
[The pKa of ketone / ester / |-keto ester is 20 / 25 / 11.]
The |-keto ester products of Claisen condensations are more acidic than simple ketones, aldehydes,
and esters because deprotonation gives an enolate whose negative charge is delocalized over both
carbonyl groups. Because |-keto esters have pKa values around 11, it is rapidly and completely
deprotonated. After the reaction is completed, addition of dilute acid converts the enolate back to the
|-keto ester.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
407
Exercise: show how the following |-keto ester could be made from Claisen condensation.
22.13 The Dieckmann condensation (intramolecular Claisen condensation)
Similar to intramolecular aldol condensation (aldol cyclization), Claisen condensation could be
carried out intramolecularly to from a ring (a ring having the functionality of |-keto ester, cyclic
|-keto ester ). This approach is called Dieckmann condensation or Dieckmann cyclization.
5-membered cyclic |-keto ester
6-membered cyclic |-keto ester
22.14 Crossed Claisen condensation
Similar to crossed aldol condensation, Claisen condensation could be carried out between different
esters. However, one of the ester without o hydrogens is necessary. Here are some useful esters
without o hydrogens are benzoate, fromate, carbonate, and oxalate esters.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
408
A successful crossed Claisen condensation is carried out by first adding the ester without o
hydrogens to s solution of the alkoxide base. The ester with o hydrogens is slowly added to this
solution, where it forms an enolate and condenses.
22.15 Syntheses using |-dicarbonyl compounds
Most ester condensation use alkoxides to from enolate ions. With simple esters, only a small amount
of enolate if formed. The equilibrium favors the alkoxide and the ester. The alkoxide often interferes
the reactions. For example: the alkoxide ion in the solution will attack the alkyl halide and form an
ester.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
409
In contrast, |-dicarbonyl compounds, such as malonic ester and acetoacetic ester, are more acidic
than alcohols. They are completely deportonated by alkoxides, and the resulting enolate are easily
alkylated and acylated. The enhanced acidity results form increased stability of the enolate ion. The
negative charge is delocalized through the resonance structures.
22.16/22.17 The malonic ester synthesis/ acetoacetic ester synthesis
The malonic ester synthesis makes substituted derivatives of acetic acid, where the acetoacetic ester
synthesis is used to make substituted derivative acetone.
The malonic ester synthesis (substituted acetic acid)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
410
Mechansim
Step 1: alkylation
Step 2: hydrolysis
Step 3: decarboxylation (|-keto acid)
The acetoacetic ester synthesis (substituted acetone)
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
411
Plan a synthesis for each of the following compound
1)
2)
22.18 Conjugated addition: The Michael reaction
o,|-Unsaturated carbonyl compounds have two electrophilic sites for nucleophile attacking. A
nucleophilie would like to attack at either the carbonyl group or at the |-position.
When attack occurs at the carbonyl group, protonation of the oxygen leads to a 1,2-addition product.
When attack occurs at the |-position, the the product, is called a 1,4-addition product. Sometimes
we called congujated addition. Conjugated addition of a carbanion to the double bond of an
o,|-unsaturated carbonyl compound is called a Michael addition. Other nucleophiles (rather than
carbon nucleophile) is still called conjugated addition.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
412
Some of the Michanel acceptors or Michael donors are shown in the following table.
Michael additions are useful in acetoacetic ester synthesis and malonic ester synthesis. Consider
the addition of the malonic ester enolate to methyl vinyl ketone (MVK). From the points of view of
organic chemist, it provides a effective method to make 1,5-dicarbonyl compounds.
Mechanism
Step 1: Michael addition to MVK.
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
413
Step 2: hydrolysis to make 1,5-dicarbonyl compound.
Exercise: how to make this compound with 1,5-dicarbonyl functionality? (hint: Stork)
22.19 The Robinson annulations (the last section)
When the compound, shown above, is treated strongly basic or acidic conditions, this 1,5-dicarbonyl
compound will undergo intramolecular aldol condensation, usually with dehydration, to give a
new six-membered ring: a conjugated cyclohexnone. This synthesis is called the Robinson
annulation. Annulation means ring formation.
Robinson annulation =Michael addition +intramolecular aldo condensation.
The mechanism
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
414
Exercise: Propose a mechanism for the following reaction:
These strategies (one reaction followed by another reaction) are often used in organic synthesis.
These types of reactions are called domino reactions or tandem reactions. A nice documentary
book is:
L.-F. Tietze, Domino reactions in organic synthesis, Wiley, Weiheim, 2006.
Go on to the next page!
Dept.Chem.FuJenUniversity OrganicChemistryA(onlyforeducation) CheChienChang
415
Suggesting reading in organic synthesis
(a) S. Warren; P. Wyatt, Organic synthesis: The disconnection approach, 2
nd
Edition, Wiley, 2008.
(b) G. S. Zweifel; M. H. Nantz, Modern Organic Synthesis: an introduction, Freeman, 2007.
Suggesting reading in organic analysis
(a) R. M. Silverstein, F. X. Webster, D. J . Kiemle, Spectrometric Identification of Organic
Compounds, Wiley, 2005.
If you have any problems about organic chemistry in the future, you can come to me at CH 227 or
CH318 as long as you are a student in this department. Good luck!
THE END OF THIS CLASS
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5822)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Mpemba Effect When Can Hot Water Freeze Faster Than ColdDocument10 pagesThe Mpemba Effect When Can Hot Water Freeze Faster Than Cold陳琮方No ratings yet
- Assignment 10Document3 pagesAssignment 10Ayush shuklaNo ratings yet
- Total HardnessDocument6 pagesTotal HardnesspermatakomputerNo ratings yet
- CS095 Membrane ElectrolyzerDocument13 pagesCS095 Membrane ElectrolyzerJuan Diego Arbelaez AlzateNo ratings yet
- 04 (2) HJBHDocument3 pages04 (2) HJBHncubepharmaNo ratings yet
- Explanation SnowDocument2 pagesExplanation SnowAhmad RamadanNo ratings yet
- Mse HWDocument2 pagesMse HWJessica AguilarNo ratings yet
- Written Exam Supramolecular Chemistry Winter 2018 F. Diederich, Y. YamakoshiDocument5 pagesWritten Exam Supramolecular Chemistry Winter 2018 F. Diederich, Y. YamakoshiSelim RezaNo ratings yet
- Student Guidance Cell-NITC Chemistry QuestionsDocument5 pagesStudent Guidance Cell-NITC Chemistry QuestionsVedurupaka Venkata SaiNo ratings yet
- Good Paper!!!Document9 pagesGood Paper!!!muriloinnocentiniNo ratings yet
- Essentials of Heat and Fluid Flow in Porous Media: PrefaceDocument3 pagesEssentials of Heat and Fluid Flow in Porous Media: PrefaceAbdul Rauf AttariNo ratings yet
- Olga Afa PDFDocument91 pagesOlga Afa PDFSofiane BouradaNo ratings yet
- DiffusionDocument2 pagesDiffusionKrishna Kumar MishraNo ratings yet
- C H E M 1 0 1: Chem 101-General Chemistry Energy & ChemistryDocument46 pagesC H E M 1 0 1: Chem 101-General Chemistry Energy & ChemistryRyo SumidaNo ratings yet
- USP-NF Atorvastatin Calcium TabletsDocument9 pagesUSP-NF Atorvastatin Calcium TabletsPhạm Đức LộcNo ratings yet
- Faraday's Law On ElectrolysisDocument2 pagesFaraday's Law On ElectrolysisFavourNo ratings yet
- Methoxyphenol (Although 3-Allyl-4-Methoxyphenol Can't Be Excluded Either) - Thus, The ProtonDocument5 pagesMethoxyphenol (Although 3-Allyl-4-Methoxyphenol Can't Be Excluded Either) - Thus, The ProtonAnonymous f8P42082UNo ratings yet
- Salt Analysis Notes 12Document42 pagesSalt Analysis Notes 12allancholan200609No ratings yet
- Measurement Systems: Application and Design by Ernest O. DoebelinDocument19 pagesMeasurement Systems: Application and Design by Ernest O. DoebelinAlhji AhmedNo ratings yet
- Adnan Aljarallah 1988 Kinetic of MTBE Over AmberlystDocument6 pagesAdnan Aljarallah 1988 Kinetic of MTBE Over AmberlystJason NunezNo ratings yet
- Introduction To Power PlantDocument31 pagesIntroduction To Power PlantLofi Radio100% (1)
- UV-Vis Application - Quantitative Analysis Using Second-Order Derivative Spectrum No A349Document2 pagesUV-Vis Application - Quantitative Analysis Using Second-Order Derivative Spectrum No A349Ramon Trinidad De la ONo ratings yet
- Introduction To ChemistryDocument116 pagesIntroduction To ChemistryJohn Michael SomorostroNo ratings yet
- Water Conveyance With Syphons: September, 2000 (Rev 2009)Document19 pagesWater Conveyance With Syphons: September, 2000 (Rev 2009)Sameer ShrivastavaNo ratings yet
- Silicon Photonics: ApplicationsDocument7 pagesSilicon Photonics: ApplicationscadmoscelticNo ratings yet
- Detailed Lesson Plan in ScienceDocument4 pagesDetailed Lesson Plan in ScienceAdrian TastarNo ratings yet
- Dapus TerbaruDocument4 pagesDapus TerbaruElvina iskandarNo ratings yet
- Astm D6038 - 2014Document4 pagesAstm D6038 - 2014alferedNo ratings yet
- Principles of Food and Bioprocess Engineering (FS 231) Problems On Heat TransferDocument8 pagesPrinciples of Food and Bioprocess Engineering (FS 231) Problems On Heat Transferx비No ratings yet
- Chapter 2-Atomic Structure Worksheet AnswersDocument2 pagesChapter 2-Atomic Structure Worksheet AnswershomamunfatNo ratings yet