Download as pdf or txt
You might also like
- Lecture 16 Introduction To Law & Ethics in Dentistry PDFDocument28 pagesLecture 16 Introduction To Law & Ethics in Dentistry PDFبروف فى اللغة الإنجليزية100% (2)
- Evaluating Cell Fractionation Procedures and Cytochemical TestsDocument6 pagesEvaluating Cell Fractionation Procedures and Cytochemical TestsJosh Zollman100% (1)
- Biochemistry II End of Semester Exam QuestionsDocument6 pagesBiochemistry II End of Semester Exam Questionsnima00No ratings yet
- Experiments in Molecular Cell Biology: A Problems Book With Multiple-Choice Question-Based TestsDocument20 pagesExperiments in Molecular Cell Biology: A Problems Book With Multiple-Choice Question-Based TestsKudumarNo ratings yet
- Respiratory Air Flow and Volume PDFDocument5 pagesRespiratory Air Flow and Volume PDFKim Robertson0% (1)
- Clinical Embryology Leeds AdsDocument1 pageClinical Embryology Leeds AdsWael BadranNo ratings yet
- Post Translational ModificationsDocument35 pagesPost Translational ModificationsGandhiraj VijayaragavanNo ratings yet
- Lecture 3 - Chapter 8-Cytoskeleton ADocument75 pagesLecture 3 - Chapter 8-Cytoskeleton AKw Chan33% (3)
- Protein FunctionDocument39 pagesProtein FunctionDeana Namirembe100% (1)
- CytoskeletonDocument86 pagesCytoskeletonAbid Al RezaNo ratings yet
- Prof Budi Wiweko - Protokol Dan Pemantauan Stimulasi Ovarium SederhanaDocument36 pagesProf Budi Wiweko - Protokol Dan Pemantauan Stimulasi Ovarium SederhanaAnisanang BanjarNo ratings yet
- Teknik Analisis Biologi Moleku PDFDocument40 pagesTeknik Analisis Biologi Moleku PDFACHMAD HAIRIL AffanNo ratings yet
- SOAL Masuk 2018Document142 pagesSOAL Masuk 2018Yoza FirdaozNo ratings yet
- Gene ExpressionDocument23 pagesGene ExpressionSutama ArtaNo ratings yet
- Genes and GenomesDocument66 pagesGenes and Genomesselowest100% (1)
- Cell Biology LabManual Version 8.0 May 2012 1 TRUNCATEDDocument57 pagesCell Biology LabManual Version 8.0 May 2012 1 TRUNCATEDதுர்காஸ்ரீ கங்கா ராதிகாNo ratings yet
- Genetics of Cystic FibrosisDocument3 pagesGenetics of Cystic Fibrosislutfiah_liaNo ratings yet
- Konsep Dasar Genetika KedokteranDocument40 pagesKonsep Dasar Genetika KedokteranSarah Nadia RasidiNo ratings yet
- PCR Basics: Kanadi SumaprajaDocument31 pagesPCR Basics: Kanadi SumaprajaSamrichardNo ratings yet
- Embryology HistoryDocument18 pagesEmbryology HistoryMelissa Monique Peart-Yates100% (1)
- Euchromatin and Heterochromatin PDFDocument2 pagesEuchromatin and Heterochromatin PDFRachelNo ratings yet
- Kelainan Kongenital Muskuloskeletal: Dr.H.Ahmad Rizal Spb. Spbo. FicsDocument35 pagesKelainan Kongenital Muskuloskeletal: Dr.H.Ahmad Rizal Spb. Spbo. Ficsranti100% (1)
- ABDocument22 pagesABpk 77No ratings yet
- Animal Models in Orthopedic Research: The Proper Animal Model To Answer Fundamental Questions On Bone Healing Depending On Pathology and Implant MaterialDocument19 pagesAnimal Models in Orthopedic Research: The Proper Animal Model To Answer Fundamental Questions On Bone Healing Depending On Pathology and Implant MaterialidobonnNo ratings yet
- Prinsip BiologiDocument67 pagesPrinsip BiologiBambang IrwansyahNo ratings yet
- Seven Jump Dalam Diskusi TutorialDocument39 pagesSeven Jump Dalam Diskusi TutorialkadinfathiaNo ratings yet
- Protein StructureDocument41 pagesProtein Structureteklay100% (1)
- ObsgynDocument1 pageObsgynaris hariyantoNo ratings yet
- Genetic Aspect of Folliculogenesis: Budi WiwekoDocument72 pagesGenetic Aspect of Folliculogenesis: Budi WiwekoAgusNo ratings yet
- 15) Dr. Firdaus, Ph. D - Penjelasan Praktikum REPRODUKSI PALUDocument35 pages15) Dr. Firdaus, Ph. D - Penjelasan Praktikum REPRODUKSI PALUcimyNo ratings yet
- Signal Transduction: Key TopicsDocument39 pagesSignal Transduction: Key TopicsBellony Sanders100% (1)
- Factors Affecting Microbial Growth in VitroDocument4 pagesFactors Affecting Microbial Growth in VitroHarline GonzagaNo ratings yet
- Vitrification ManualDocument46 pagesVitrification Manualjose.antoni3480No ratings yet
- Biochemical Energetics: Peranan Atp BioenergetikaDocument23 pagesBiochemical Energetics: Peranan Atp BioenergetikaHerryNo ratings yet
- Third Week of DevelopmentDocument34 pagesThird Week of DevelopmentAriba Asif100% (1)
- Compilation of Oxford Q&ADocument16 pagesCompilation of Oxford Q&AAdmin DutiesNo ratings yet
- Whole Genome Sequencing of HumanDocument17 pagesWhole Genome Sequencing of Humanbiovijay101No ratings yet
- Cervical Cancer Referral PathwayDocument1 pageCervical Cancer Referral PathwaySifa AditNo ratings yet
- Male Reproductive Function Sept 2014Document32 pagesMale Reproductive Function Sept 2014Nirmesh ThanabalanNo ratings yet
- Limb DevelopmentDocument95 pagesLimb Developmentcandy_kb17100% (1)
- Biotechnology SyllabusDocument111 pagesBiotechnology SyllabusSatyam SinghNo ratings yet
- 2012 Utrogestan Natural Micronized Progesterone - From Luteal Phase Defect To Preterm BirthDocument48 pages2012 Utrogestan Natural Micronized Progesterone - From Luteal Phase Defect To Preterm BirthRachel Paredes100% (1)
- FASTADocument33 pagesFASTAAnton MelcherNo ratings yet
- Microcytic Anemia PDFDocument8 pagesMicrocytic Anemia PDFMargarita TorresNo ratings yet
- Jurnal Bronchitis Dengan AsmaDocument5 pagesJurnal Bronchitis Dengan AsmaMauLan SaputraNo ratings yet
- Biology of Green Sulfur BacteriaDocument12 pagesBiology of Green Sulfur BacteriaOswaldo EncisoNo ratings yet
- Illumina IVF BrochureDocument12 pagesIllumina IVF BrochureEmily KongNo ratings yet
- CryopreservationDocument29 pagesCryopreservationDharmendra KumarNo ratings yet
- Facts About The Human GenomeDocument14 pagesFacts About The Human Genomeapi-19734323No ratings yet
- Cloning Dolly & MicromanipulationDocument29 pagesCloning Dolly & Micromanipulationnitinyadav16No ratings yet
- PROTEOMICSDocument18 pagesPROTEOMICSRashmi YadavNo ratings yet
- MSC MPlil ZoologyDocument18 pagesMSC MPlil ZoologyYougesh KumarNo ratings yet
- PCR An OverviewDocument41 pagesPCR An OverviewnassradabourNo ratings yet
- Pembelahan Embrio (Cleavage) S2 2017Document47 pagesPembelahan Embrio (Cleavage) S2 2017Desy NataliaNo ratings yet
- Embryology - Updates and Highlights On Classic TopicsDocument222 pagesEmbryology - Updates and Highlights On Classic TopicsIndera Vyas100% (1)
- Polycistronic and Monocistronic mRNAsDocument7 pagesPolycistronic and Monocistronic mRNAskailas ambadi100% (1)
- Genomics: A New Revolution in Science:: An Introduction To Promises and Ethical Considerations by Genome AlbertaDocument66 pagesGenomics: A New Revolution in Science:: An Introduction To Promises and Ethical Considerations by Genome AlbertaKaren LowNo ratings yet
- BIO 516-Advance Molecular Biology-Shaper MirzaDocument4 pagesBIO 516-Advance Molecular Biology-Shaper MirzaAnonymous sF8ZuiGNo ratings yet
- Deep Brain StimulationDocument4 pagesDeep Brain StimulationCristian GondacNo ratings yet
- Genome OrganizationDocument11 pagesGenome OrganizationKakahsa KhanNo ratings yet
- Tension Pneumothorax PDFDocument2 pagesTension Pneumothorax PDFClarissa Aileen Caliva AdoraNo ratings yet
- Morpheus8 Consent FormDocument3 pagesMorpheus8 Consent FormOnurNo ratings yet
- ENG Paper 2 TrialDocument17 pagesENG Paper 2 TrialstudentmbsNo ratings yet
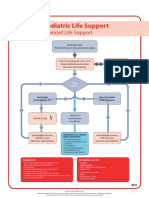
- Poster 10 PALS 01 01 ENG V20100927 PDFDocument1 pagePoster 10 PALS 01 01 ENG V20100927 PDFAndy XiaoNo ratings yet
- Solid Dosage Forms in Unani System of Medicine AnDocument7 pagesSolid Dosage Forms in Unani System of Medicine AnSiddharth JainNo ratings yet
- mg359 All PDFDocument24 pagesmg359 All PDFsyedsrahmanNo ratings yet
- BloodlettingDocument5 pagesBloodlettingMaria Lana Grace DiazNo ratings yet
- Difference Between Genetic and Non Genetic RnaDocument1 pageDifference Between Genetic and Non Genetic RnaMatin Ahmad KhanNo ratings yet
- Preventive Care: Female Checklist - UnitedHealthcareDocument3 pagesPreventive Care: Female Checklist - UnitedHealthcareLiz MNo ratings yet
- NewEarth - University COVID-19 Intelligence BriefDocument47 pagesNewEarth - University COVID-19 Intelligence BriefTony Lambert100% (1)
- Trauma and Burns MCQDocument74 pagesTrauma and Burns MCQAhmed Kassem100% (2)
- Isolation GuidelinesDocument203 pagesIsolation GuidelinesAlifia TrisnaningrumNo ratings yet
- 02/27/09 - The Stanford Daily (PDF)Document8 pages02/27/09 - The Stanford Daily (PDF)The Stanford DailyNo ratings yet
- Transportation in AnimalsDocument14 pagesTransportation in AnimalsCREATIVING TAMILANNo ratings yet
- Vital Sign #1: Body TemperatureDocument5 pagesVital Sign #1: Body TemperaturePrimelift Safety Resources LimitedNo ratings yet
- Additional Mathematics Project 2012 EXAMPLEDocument22 pagesAdditional Mathematics Project 2012 EXAMPLEAmanda F.100% (1)
- Lecture 5 Birth DefectsDocument13 pagesLecture 5 Birth DefectsDenis KaguiNo ratings yet
- Office Work HiraDocument2 pagesOffice Work HiratirthNo ratings yet
- Posterior Reversible Encephalopathy Syndrome in A Postpartum Normotensive Post Cesarean Patient - A Case ReportDocument4 pagesPosterior Reversible Encephalopathy Syndrome in A Postpartum Normotensive Post Cesarean Patient - A Case ReportIJAR JOURNALNo ratings yet
- Diagnosis and Correction of Uterine Torsion in Cattle and BuffaloesDocument13 pagesDiagnosis and Correction of Uterine Torsion in Cattle and Buffaloesgnpoba100% (12)
- Day 1 General Objective:: CognitiveDocument2 pagesDay 1 General Objective:: CognitiveReihann N. EdresNo ratings yet
- Hoërskool Roodepoort: COVID-19 PolicyDocument24 pagesHoërskool Roodepoort: COVID-19 PolicyScarfacembali MbaliNo ratings yet
- Supliment-2022 RJC PDFDocument320 pagesSupliment-2022 RJC PDFSimona IonitaNo ratings yet
- Tues 10-20 Peripheral Nerve Disorders - A Practical OverviewDocument36 pagesTues 10-20 Peripheral Nerve Disorders - A Practical OverviewGery FirmansyahNo ratings yet
- Vascular DementiaDocument57 pagesVascular Dementiadrkadiyala2No ratings yet
- Nelec 2: Acute and Critical Care Nursing: Unit Learning OutcomeDocument4 pagesNelec 2: Acute and Critical Care Nursing: Unit Learning Outcomeyas minNo ratings yet
- Incorporating Mindfulness Meditation Into The Treatment of Provoked VestibulodyniaDocument10 pagesIncorporating Mindfulness Meditation Into The Treatment of Provoked VestibulodyniaPau VRNo ratings yet
- Antibacterial HydrogelsDocument17 pagesAntibacterial HydrogelsALONDRA CAROLINA HERNANDEZ QUINTERONo ratings yet
- Republic v. Cagandahan, G.R. No. 166676, 12 September 2008Document1 pageRepublic v. Cagandahan, G.R. No. 166676, 12 September 2008Jovelan V. EscañoNo ratings yet