
8 Suarez
8 Suarez
You might also like
- Metallic Oxides by GoodenoughDocument255 pagesMetallic Oxides by Goodenoughmuk_hawkNo ratings yet
- Aerzen Vibration StandardDocument3 pagesAerzen Vibration StandardFirdausrosli84No ratings yet
- Ater Chillers: Euro ChillerDocument8 pagesAter Chillers: Euro ChillerAnonymous lB6SsHu5KNo ratings yet
- MECH4406 HeatTransfer Outline F2018 Rev2Document3 pagesMECH4406 HeatTransfer Outline F2018 Rev2Ryan HiraniNo ratings yet
- Theoretical Study of Electronic, Magnetic, and Structural Properties of α-Fe2O3 (Hematite)Document11 pagesTheoretical Study of Electronic, Magnetic, and Structural Properties of α-Fe2O3 (Hematite)Eder Ruiz HernandezNo ratings yet
- 8.6 Spinel, Perovskite, and Rutile Structures PDFDocument5 pages8.6 Spinel, Perovskite, and Rutile Structures PDFΑντώνης ΜακρίδηςNo ratings yet
- L. Mathey, S. - W. Tsai and A. H. Castro Neto - Exotic Superconducting Phases of Ultracold Atom Mixtures On Triangular LatticesDocument7 pagesL. Mathey, S. - W. Tsai and A. H. Castro Neto - Exotic Superconducting Phases of Ultracold Atom Mixtures On Triangular LatticesItama23No ratings yet
- Alexander Yaresko - Magnetic Properties of Iron Pnictides From Spin-Spiral CalculationsDocument4 pagesAlexander Yaresko - Magnetic Properties of Iron Pnictides From Spin-Spiral CalculationsPo48HSDNo ratings yet
- Oxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3Document23 pagesOxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3solisiusNo ratings yet
- Structural and Magnetic Phase Diagram of Cefeaso F and Its Relationship To High-Temperature SuperconductivityDocument19 pagesStructural and Magnetic Phase Diagram of Cefeaso F and Its Relationship To High-Temperature SuperconductivityPoemMenungguPagiNo ratings yet
- Rkky Ferromagnetism With Ising-Like Spin States in Intercalated Fe TasDocument5 pagesRkky Ferromagnetism With Ising-Like Spin States in Intercalated Fe TashkphyNo ratings yet
- Mishra Chem Mater 2010Document11 pagesMishra Chem Mater 2010MemoVillasenorNo ratings yet
- Line-Shape Analysis of Optical Spectra in Metaphosphate Glasses Doped With Erbium IonsDocument9 pagesLine-Shape Analysis of Optical Spectra in Metaphosphate Glasses Doped With Erbium Ions66666666666666-35305No ratings yet
- 2020 SK Et Al JMMMDocument7 pages2020 SK Et Al JMMMsavankatbaNo ratings yet
- Effects of An Electric Eld On The Con Ned Hydrogen AtomDocument4 pagesEffects of An Electric Eld On The Con Ned Hydrogen AtomvgybhunjiNo ratings yet
- The Crystal Structure of Diamond - DawsonDocument26 pagesThe Crystal Structure of Diamond - Dawsonsal.paradise.1No ratings yet
- Electronic Structure, Magnetic and Optical Properties of Quaternary Fe Co Mnal Heusler AlloysDocument12 pagesElectronic Structure, Magnetic and Optical Properties of Quaternary Fe Co Mnal Heusler AlloysDaljot Singh KangNo ratings yet
- Parlinski sfn201112008 2011Document6 pagesParlinski sfn201112008 2011IvanNo ratings yet
- Long-Range Proximity Effects in Superconductor-Ferromagnet StructuresDocument4 pagesLong-Range Proximity Effects in Superconductor-Ferromagnet StructurespanzhanghiggsNo ratings yet
- Crystal Structure of Na (Moo) (P O) Studied by Synchrotron X-Ray DiffractionDocument5 pagesCrystal Structure of Na (Moo) (P O) Studied by Synchrotron X-Ray DiffractionMiguel Ángel Laguna MartínezNo ratings yet
- Ismail A. M. Ibrahim, Zoltán Lenčéš, Pavol Šajgalík, Lubomir Benco, Martijn MarsmanDocument23 pagesIsmail A. M. Ibrahim, Zoltán Lenčéš, Pavol Šajgalík, Lubomir Benco, Martijn MarsmanashNo ratings yet
- General Static Polarizability in Spherical Neutral Metal Clusters and Fullerenes Within Thomas-Fermi TheoryDocument17 pagesGeneral Static Polarizability in Spherical Neutral Metal Clusters and Fullerenes Within Thomas-Fermi TheoryThomas PaladeNo ratings yet
- Inhomogeneity-Induced High Temperature Ferromagnetism in N-Type Ferromagnetic Semiconductor (In, Fe) As Grown On Vicinal Gaas SubstratesDocument9 pagesInhomogeneity-Induced High Temperature Ferromagnetism in N-Type Ferromagnetic Semiconductor (In, Fe) As Grown On Vicinal Gaas Substratesparra MedinaNo ratings yet
- Bi-Mn Doped SmFeO3 NancyDocument99 pagesBi-Mn Doped SmFeO3 NancyRAJABLA NAINNo ratings yet
- Michaela TU Ková, Ji Í Tu EK, Pavel TU EK, Lubomír KUBÁ EKDocument6 pagesMichaela TU Ková, Ji Í Tu EK, Pavel TU EK, Lubomír KUBÁ EKFaouzi TlemcenNo ratings yet
- Enhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryDocument3 pagesEnhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryJuaxmawNo ratings yet
- Atomic Recon Figuration Among Tri-State Transition at Ferroelectric/antiferroelectric Phase Boundaries in PB (ZR, Ti) ODocument9 pagesAtomic Recon Figuration Among Tri-State Transition at Ferroelectric/antiferroelectric Phase Boundaries in PB (ZR, Ti) OAnh Tuấn TrầnNo ratings yet
- Asymmetry in Mössbauer Spectra of Fe - Azaporphyrin ComplexesDocument6 pagesAsymmetry in Mössbauer Spectra of Fe - Azaporphyrin ComplexesGudo SidhuNo ratings yet
- Effect of Yttrium Substitution On The Structural and Magnetic Properties of Smfeo3Document5 pagesEffect of Yttrium Substitution On The Structural and Magnetic Properties of Smfeo3rautsubhajit89No ratings yet
- Zhang 2010Document4 pagesZhang 2010Wenyo ChenNo ratings yet
- CCTODocument8 pagesCCTOjakharnarenNo ratings yet
- Ab Initio Analysis of Metal-Ligand Bonding in An (COT) 2 WithDocument14 pagesAb Initio Analysis of Metal-Ligand Bonding in An (COT) 2 WithrafelNo ratings yet
- First Principle StudyDocument20 pagesFirst Principle StudyabdallaNo ratings yet
- First Principles Calculations of Structural, Electronic and Magnetic Properties of MN Based Magnetic Semiconductor Al MN SBDocument11 pagesFirst Principles Calculations of Structural, Electronic and Magnetic Properties of MN Based Magnetic Semiconductor Al MN SBimprincesssNo ratings yet
- 1 s2.0 S0167577X15008368 MainDocument4 pages1 s2.0 S0167577X15008368 MainHuckkey HuNo ratings yet
- Ballistic Transfer of Spin Momentum in Multiferroic Tunnel JunctionDocument4 pagesBallistic Transfer of Spin Momentum in Multiferroic Tunnel JunctionglebdmirichNo ratings yet
- Monoelectronic Band Structure For A Cubic PerovskiteDocument1 pageMonoelectronic Band Structure For A Cubic PerovskiteSJNo ratings yet
- 10 1103@PhysRevB 94 165419Document10 pages10 1103@PhysRevB 94 165419Carlos PaezNo ratings yet
- Hisao Kobayashi Et Al - Phonon Density of States and Compression Behavior in Iron Sulfide Under PressureDocument4 pagesHisao Kobayashi Et Al - Phonon Density of States and Compression Behavior in Iron Sulfide Under PressureDrebuioNo ratings yet
- Crystals 14 00299Document20 pagesCrystals 14 00299jamel-shamsNo ratings yet
- Cationic Exchange in Nanosized Znfe O Spinel Revealed by Experimental and Simulated Near-Edge Absorption StructureDocument5 pagesCationic Exchange in Nanosized Znfe O Spinel Revealed by Experimental and Simulated Near-Edge Absorption StructureAna Paula FreitasNo ratings yet
- ECE1006 Introduction To Nanoscience and Nanotechnology Digital Assignment 1Document7 pagesECE1006 Introduction To Nanoscience and Nanotechnology Digital Assignment 1Karthik ReddyNo ratings yet
- A Comparative Study of Nanosized Iron Oxide ParticlesDocument7 pagesA Comparative Study of Nanosized Iron Oxide Particlessastrika aninditaNo ratings yet
- Previous Lecture: (And On Shape, Size, Neighbors)Document16 pagesPrevious Lecture: (And On Shape, Size, Neighbors)idownloadbooks3133No ratings yet
- Magnetic Structure of Molecular Magnet Fe (Fe (CN) ) 4H ODocument6 pagesMagnetic Structure of Molecular Magnet Fe (Fe (CN) ) 4H ONarayanraj RajNo ratings yet
- Imaging Molecular Geometry With Electron Momentum SpectrosDocument8 pagesImaging Molecular Geometry With Electron Momentum SpectrosFilip KesteliNo ratings yet
- Surface Plasmon Polaritons On Metallic Surfaces: A. R. Zakharian, J. V. Moloney, and M. MansuripurDocument15 pagesSurface Plasmon Polaritons On Metallic Surfaces: A. R. Zakharian, J. V. Moloney, and M. Mansuripuranon_489850512No ratings yet
- MagnetiteFe3O4111Surfaces Si 001Document11 pagesMagnetiteFe3O4111Surfaces Si 001Deni haryadiNo ratings yet
- Orbital Physics in Transition-Metal Oxides From First-PrinciplesDocument8 pagesOrbital Physics in Transition-Metal Oxides From First-PrinciplesalfonsoNo ratings yet
- Crystal Structures and Electrical Conducting/Magnetic Properties in 1: 1 Fecl and Febr Salts of Dimethylthio-And Ethylenedithio - (1,3 - Dithiolylidene) Thioxotetrathiafulvalene Radical CationsDocument6 pagesCrystal Structures and Electrical Conducting/Magnetic Properties in 1: 1 Fecl and Febr Salts of Dimethylthio-And Ethylenedithio - (1,3 - Dithiolylidene) Thioxotetrathiafulvalene Radical CationsJagajith Sathis Chandran NairNo ratings yet
- Introduction To Molecular Orbital TheoryDocument56 pagesIntroduction To Molecular Orbital TheorylastlegendNo ratings yet
- Epr NotesDocument40 pagesEpr NotesArunachalamNo ratings yet
- Magnetochemistry 08 00144Document30 pagesMagnetochemistry 08 00144solisiusNo ratings yet
- Investigation of Spin Reorientation in Ymn1-Xfexo3 (X 0.55, 0.6, 0.7, 0.8, 0.9, and 1.0) by Mössbauer SpectrosDocument4 pagesInvestigation of Spin Reorientation in Ymn1-Xfexo3 (X 0.55, 0.6, 0.7, 0.8, 0.9, and 1.0) by Mössbauer Spectrosrautsubhajit89No ratings yet
- Structure Properties of Liquid FCC Transition Metals Using The Embedded Atom Method PotentialDocument8 pagesStructure Properties of Liquid FCC Transition Metals Using The Embedded Atom Method PotentialivasitonNo ratings yet
- Contribution Presented at NAPP'03, May 26-31, 2003, Dubrovnik, CroatiaDocument10 pagesContribution Presented at NAPP'03, May 26-31, 2003, Dubrovnik, CroatiameNo ratings yet
- Investigation of Superconducting Gap Structure in Hfirsi Using Muon Spin Relaxation/RotationDocument9 pagesInvestigation of Superconducting Gap Structure in Hfirsi Using Muon Spin Relaxation/RotationkartikNo ratings yet
- Real DFT Model On Co Adsorption by Noble CatalystDocument52 pagesReal DFT Model On Co Adsorption by Noble CatalystDikra BkNo ratings yet
- Enhancement of Thermoelectric Properties of Porphyrin-Based 2021Document7 pagesEnhancement of Thermoelectric Properties of Porphyrin-Based 2021Toshar GargNo ratings yet
- 1 s2.0 S0277538721003818 MainDocument11 pages1 s2.0 S0277538721003818 MainMoromi NathNo ratings yet
- Ja 1Document15 pagesJa 1titrantNo ratings yet
- Amorphous Semiconductors: Structural, Optical, and Electronic PropertiesFrom EverandAmorphous Semiconductors: Structural, Optical, and Electronic PropertiesNo ratings yet
- Computational Methods in Lanthanide and Actinide ChemistryFrom EverandComputational Methods in Lanthanide and Actinide ChemistryMichael DolgNo ratings yet
- Settlement Analysis (Two Way Drainage)Document5 pagesSettlement Analysis (Two Way Drainage)Tanmoy DasNo ratings yet
- Pile Analysis, Design and DetailingDocument15 pagesPile Analysis, Design and DetailingMovies PointNo ratings yet
- DL2250,3000,4500,6000 PDFDocument138 pagesDL2250,3000,4500,6000 PDFPedro Pablo GonzalezNo ratings yet
- Procedure: Activity 2: Where in The World Is The Philippines? Part IiDocument28 pagesProcedure: Activity 2: Where in The World Is The Philippines? Part Iisamn cadNo ratings yet
- Radiation Protection & Quality Assurance in CT 2018Document51 pagesRadiation Protection & Quality Assurance in CT 2018abafzNo ratings yet
- W1 B Plane - Strain - TransformationDocument29 pagesW1 B Plane - Strain - TransformationSafwan RawiNo ratings yet
- Lesson+5+ +air Conditioning+and+Indoor+Air+QualityDocument53 pagesLesson+5+ +air Conditioning+and+Indoor+Air+QualityAmelia Isabelle VilagaNo ratings yet
- MCQ Chapter 2 ElectrochemistryDocument4 pagesMCQ Chapter 2 ElectrochemistryFact GamerNo ratings yet
- 5th Notes SST Revised FormatDocument5 pages5th Notes SST Revised FormatManzoor AlamNo ratings yet
- Dynamic Analysis of Pile Foundations Bem-Fem ModelDocument14 pagesDynamic Analysis of Pile Foundations Bem-Fem ModelShravan KumarNo ratings yet
- Product Group Industrial Vacuum Cleaners, 0415-156Document8 pagesProduct Group Industrial Vacuum Cleaners, 0415-156هبال حمزةNo ratings yet
- 2018-Effect of PVA On Mechanical Properties of Tapioca Starch Reinforced Coconut Fiber Biopolymer Composite PDFDocument5 pages2018-Effect of PVA On Mechanical Properties of Tapioca Starch Reinforced Coconut Fiber Biopolymer Composite PDFSlim ShaddysNo ratings yet
- Anchors in Cracked Concrete - Hilti SingaporeDocument16 pagesAnchors in Cracked Concrete - Hilti SingaporeOGStage2 ASNo ratings yet
- Thermodynamics 1655870560521Document26 pagesThermodynamics 1655870560521Singh DhruvNo ratings yet
- A Practical Guide To Soil-Structure Interaction: FEMA P-2091 / December 2020Document218 pagesA Practical Guide To Soil-Structure Interaction: FEMA P-2091 / December 2020Asrat Worku100% (1)
- Sample PT ProcedureDocument10 pagesSample PT ProcedureLarry Keating100% (2)
- Tool Life, Tool Wear Machinability PDFDocument9 pagesTool Life, Tool Wear Machinability PDFshivaNo ratings yet
- United States Patent (10) Patent No.: US 6,499,306 B2: Gillen (45) Date of Patent: Dec. 31, 2002Document25 pagesUnited States Patent (10) Patent No.: US 6,499,306 B2: Gillen (45) Date of Patent: Dec. 31, 2002HP ghnNo ratings yet
- Reversible Aqueous Zinc/manganese Oxide Energy Storage From Conversion ReactionsDocument7 pagesReversible Aqueous Zinc/manganese Oxide Energy Storage From Conversion ReactionsShofiNo ratings yet
- Caleffi: Maxcal Automatic Air VentDocument2 pagesCaleffi: Maxcal Automatic Air VentelmerbarrerasNo ratings yet
- Motions Class-IX (Physics) Solved Test Paper - 01Document4 pagesMotions Class-IX (Physics) Solved Test Paper - 01anshika tembhareNo ratings yet
- Type of Heat ExchangersDocument46 pagesType of Heat ExchangersguravdrNo ratings yet
- 7-Boiler - Eng - Ono SuparnoDocument14 pages7-Boiler - Eng - Ono SuparnoRaihan Rivellino AdzaniNo ratings yet
- H2 and CH4 Sorption On Cu-BTC Metal Organic Framew PDFDocument7 pagesH2 and CH4 Sorption On Cu-BTC Metal Organic Framew PDFMosllem ShahmohammadiNo ratings yet
- Aerial TriangulationDocument37 pagesAerial TriangulationRussel Clarete100% (1)
- Chapter 6 - Fatigue FailuresDocument70 pagesChapter 6 - Fatigue FailuresKanakNo ratings yet




































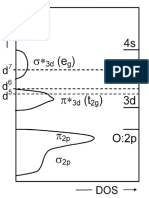



















































Download as pdf or txt
You might also like
- Metallic Oxides by GoodenoughDocument255 pagesMetallic Oxides by Goodenoughmuk_hawkNo ratings yet
- Aerzen Vibration StandardDocument3 pagesAerzen Vibration StandardFirdausrosli84No ratings yet
- Ater Chillers: Euro ChillerDocument8 pagesAter Chillers: Euro ChillerAnonymous lB6SsHu5KNo ratings yet
- MECH4406 HeatTransfer Outline F2018 Rev2Document3 pagesMECH4406 HeatTransfer Outline F2018 Rev2Ryan HiraniNo ratings yet
- Theoretical Study of Electronic, Magnetic, and Structural Properties of α-Fe2O3 (Hematite)Document11 pagesTheoretical Study of Electronic, Magnetic, and Structural Properties of α-Fe2O3 (Hematite)Eder Ruiz HernandezNo ratings yet
- 8.6 Spinel, Perovskite, and Rutile Structures PDFDocument5 pages8.6 Spinel, Perovskite, and Rutile Structures PDFΑντώνης ΜακρίδηςNo ratings yet
- L. Mathey, S. - W. Tsai and A. H. Castro Neto - Exotic Superconducting Phases of Ultracold Atom Mixtures On Triangular LatticesDocument7 pagesL. Mathey, S. - W. Tsai and A. H. Castro Neto - Exotic Superconducting Phases of Ultracold Atom Mixtures On Triangular LatticesItama23No ratings yet
- Alexander Yaresko - Magnetic Properties of Iron Pnictides From Spin-Spiral CalculationsDocument4 pagesAlexander Yaresko - Magnetic Properties of Iron Pnictides From Spin-Spiral CalculationsPo48HSDNo ratings yet
- Oxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3Document23 pagesOxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3solisiusNo ratings yet
- Structural and Magnetic Phase Diagram of Cefeaso F and Its Relationship To High-Temperature SuperconductivityDocument19 pagesStructural and Magnetic Phase Diagram of Cefeaso F and Its Relationship To High-Temperature SuperconductivityPoemMenungguPagiNo ratings yet
- Rkky Ferromagnetism With Ising-Like Spin States in Intercalated Fe TasDocument5 pagesRkky Ferromagnetism With Ising-Like Spin States in Intercalated Fe TashkphyNo ratings yet
- Mishra Chem Mater 2010Document11 pagesMishra Chem Mater 2010MemoVillasenorNo ratings yet
- Line-Shape Analysis of Optical Spectra in Metaphosphate Glasses Doped With Erbium IonsDocument9 pagesLine-Shape Analysis of Optical Spectra in Metaphosphate Glasses Doped With Erbium Ions66666666666666-35305No ratings yet
- 2020 SK Et Al JMMMDocument7 pages2020 SK Et Al JMMMsavankatbaNo ratings yet
- Effects of An Electric Eld On The Con Ned Hydrogen AtomDocument4 pagesEffects of An Electric Eld On The Con Ned Hydrogen AtomvgybhunjiNo ratings yet
- The Crystal Structure of Diamond - DawsonDocument26 pagesThe Crystal Structure of Diamond - Dawsonsal.paradise.1No ratings yet
- Electronic Structure, Magnetic and Optical Properties of Quaternary Fe Co Mnal Heusler AlloysDocument12 pagesElectronic Structure, Magnetic and Optical Properties of Quaternary Fe Co Mnal Heusler AlloysDaljot Singh KangNo ratings yet
- Parlinski sfn201112008 2011Document6 pagesParlinski sfn201112008 2011IvanNo ratings yet
- Long-Range Proximity Effects in Superconductor-Ferromagnet StructuresDocument4 pagesLong-Range Proximity Effects in Superconductor-Ferromagnet StructurespanzhanghiggsNo ratings yet
- Crystal Structure of Na (Moo) (P O) Studied by Synchrotron X-Ray DiffractionDocument5 pagesCrystal Structure of Na (Moo) (P O) Studied by Synchrotron X-Ray DiffractionMiguel Ángel Laguna MartínezNo ratings yet
- Ismail A. M. Ibrahim, Zoltán Lenčéš, Pavol Šajgalík, Lubomir Benco, Martijn MarsmanDocument23 pagesIsmail A. M. Ibrahim, Zoltán Lenčéš, Pavol Šajgalík, Lubomir Benco, Martijn MarsmanashNo ratings yet
- General Static Polarizability in Spherical Neutral Metal Clusters and Fullerenes Within Thomas-Fermi TheoryDocument17 pagesGeneral Static Polarizability in Spherical Neutral Metal Clusters and Fullerenes Within Thomas-Fermi TheoryThomas PaladeNo ratings yet
- Inhomogeneity-Induced High Temperature Ferromagnetism in N-Type Ferromagnetic Semiconductor (In, Fe) As Grown On Vicinal Gaas SubstratesDocument9 pagesInhomogeneity-Induced High Temperature Ferromagnetism in N-Type Ferromagnetic Semiconductor (In, Fe) As Grown On Vicinal Gaas Substratesparra MedinaNo ratings yet
- Bi-Mn Doped SmFeO3 NancyDocument99 pagesBi-Mn Doped SmFeO3 NancyRAJABLA NAINNo ratings yet
- Michaela TU Ková, Ji Í Tu EK, Pavel TU EK, Lubomír KUBÁ EKDocument6 pagesMichaela TU Ková, Ji Í Tu EK, Pavel TU EK, Lubomír KUBÁ EKFaouzi TlemcenNo ratings yet
- Enhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryDocument3 pagesEnhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryJuaxmawNo ratings yet
- Atomic Recon Figuration Among Tri-State Transition at Ferroelectric/antiferroelectric Phase Boundaries in PB (ZR, Ti) ODocument9 pagesAtomic Recon Figuration Among Tri-State Transition at Ferroelectric/antiferroelectric Phase Boundaries in PB (ZR, Ti) OAnh Tuấn TrầnNo ratings yet
- Asymmetry in Mössbauer Spectra of Fe - Azaporphyrin ComplexesDocument6 pagesAsymmetry in Mössbauer Spectra of Fe - Azaporphyrin ComplexesGudo SidhuNo ratings yet
- Effect of Yttrium Substitution On The Structural and Magnetic Properties of Smfeo3Document5 pagesEffect of Yttrium Substitution On The Structural and Magnetic Properties of Smfeo3rautsubhajit89No ratings yet
- Zhang 2010Document4 pagesZhang 2010Wenyo ChenNo ratings yet
- CCTODocument8 pagesCCTOjakharnarenNo ratings yet
- Ab Initio Analysis of Metal-Ligand Bonding in An (COT) 2 WithDocument14 pagesAb Initio Analysis of Metal-Ligand Bonding in An (COT) 2 WithrafelNo ratings yet
- First Principle StudyDocument20 pagesFirst Principle StudyabdallaNo ratings yet
- First Principles Calculations of Structural, Electronic and Magnetic Properties of MN Based Magnetic Semiconductor Al MN SBDocument11 pagesFirst Principles Calculations of Structural, Electronic and Magnetic Properties of MN Based Magnetic Semiconductor Al MN SBimprincesssNo ratings yet
- 1 s2.0 S0167577X15008368 MainDocument4 pages1 s2.0 S0167577X15008368 MainHuckkey HuNo ratings yet
- Ballistic Transfer of Spin Momentum in Multiferroic Tunnel JunctionDocument4 pagesBallistic Transfer of Spin Momentum in Multiferroic Tunnel JunctionglebdmirichNo ratings yet
- Monoelectronic Band Structure For A Cubic PerovskiteDocument1 pageMonoelectronic Band Structure For A Cubic PerovskiteSJNo ratings yet
- 10 1103@PhysRevB 94 165419Document10 pages10 1103@PhysRevB 94 165419Carlos PaezNo ratings yet
- Hisao Kobayashi Et Al - Phonon Density of States and Compression Behavior in Iron Sulfide Under PressureDocument4 pagesHisao Kobayashi Et Al - Phonon Density of States and Compression Behavior in Iron Sulfide Under PressureDrebuioNo ratings yet
- Crystals 14 00299Document20 pagesCrystals 14 00299jamel-shamsNo ratings yet
- Cationic Exchange in Nanosized Znfe O Spinel Revealed by Experimental and Simulated Near-Edge Absorption StructureDocument5 pagesCationic Exchange in Nanosized Znfe O Spinel Revealed by Experimental and Simulated Near-Edge Absorption StructureAna Paula FreitasNo ratings yet
- ECE1006 Introduction To Nanoscience and Nanotechnology Digital Assignment 1Document7 pagesECE1006 Introduction To Nanoscience and Nanotechnology Digital Assignment 1Karthik ReddyNo ratings yet
- A Comparative Study of Nanosized Iron Oxide ParticlesDocument7 pagesA Comparative Study of Nanosized Iron Oxide Particlessastrika aninditaNo ratings yet
- Previous Lecture: (And On Shape, Size, Neighbors)Document16 pagesPrevious Lecture: (And On Shape, Size, Neighbors)idownloadbooks3133No ratings yet
- Magnetic Structure of Molecular Magnet Fe (Fe (CN) ) 4H ODocument6 pagesMagnetic Structure of Molecular Magnet Fe (Fe (CN) ) 4H ONarayanraj RajNo ratings yet
- Imaging Molecular Geometry With Electron Momentum SpectrosDocument8 pagesImaging Molecular Geometry With Electron Momentum SpectrosFilip KesteliNo ratings yet
- Surface Plasmon Polaritons On Metallic Surfaces: A. R. Zakharian, J. V. Moloney, and M. MansuripurDocument15 pagesSurface Plasmon Polaritons On Metallic Surfaces: A. R. Zakharian, J. V. Moloney, and M. Mansuripuranon_489850512No ratings yet
- MagnetiteFe3O4111Surfaces Si 001Document11 pagesMagnetiteFe3O4111Surfaces Si 001Deni haryadiNo ratings yet
- Orbital Physics in Transition-Metal Oxides From First-PrinciplesDocument8 pagesOrbital Physics in Transition-Metal Oxides From First-PrinciplesalfonsoNo ratings yet
- Crystal Structures and Electrical Conducting/Magnetic Properties in 1: 1 Fecl and Febr Salts of Dimethylthio-And Ethylenedithio - (1,3 - Dithiolylidene) Thioxotetrathiafulvalene Radical CationsDocument6 pagesCrystal Structures and Electrical Conducting/Magnetic Properties in 1: 1 Fecl and Febr Salts of Dimethylthio-And Ethylenedithio - (1,3 - Dithiolylidene) Thioxotetrathiafulvalene Radical CationsJagajith Sathis Chandran NairNo ratings yet
- Introduction To Molecular Orbital TheoryDocument56 pagesIntroduction To Molecular Orbital TheorylastlegendNo ratings yet
- Epr NotesDocument40 pagesEpr NotesArunachalamNo ratings yet
- Magnetochemistry 08 00144Document30 pagesMagnetochemistry 08 00144solisiusNo ratings yet
- Investigation of Spin Reorientation in Ymn1-Xfexo3 (X 0.55, 0.6, 0.7, 0.8, 0.9, and 1.0) by Mössbauer SpectrosDocument4 pagesInvestigation of Spin Reorientation in Ymn1-Xfexo3 (X 0.55, 0.6, 0.7, 0.8, 0.9, and 1.0) by Mössbauer Spectrosrautsubhajit89No ratings yet
- Structure Properties of Liquid FCC Transition Metals Using The Embedded Atom Method PotentialDocument8 pagesStructure Properties of Liquid FCC Transition Metals Using The Embedded Atom Method PotentialivasitonNo ratings yet
- Contribution Presented at NAPP'03, May 26-31, 2003, Dubrovnik, CroatiaDocument10 pagesContribution Presented at NAPP'03, May 26-31, 2003, Dubrovnik, CroatiameNo ratings yet
- Investigation of Superconducting Gap Structure in Hfirsi Using Muon Spin Relaxation/RotationDocument9 pagesInvestigation of Superconducting Gap Structure in Hfirsi Using Muon Spin Relaxation/RotationkartikNo ratings yet
- Real DFT Model On Co Adsorption by Noble CatalystDocument52 pagesReal DFT Model On Co Adsorption by Noble CatalystDikra BkNo ratings yet
- Enhancement of Thermoelectric Properties of Porphyrin-Based 2021Document7 pagesEnhancement of Thermoelectric Properties of Porphyrin-Based 2021Toshar GargNo ratings yet
- 1 s2.0 S0277538721003818 MainDocument11 pages1 s2.0 S0277538721003818 MainMoromi NathNo ratings yet
- Ja 1Document15 pagesJa 1titrantNo ratings yet
- Amorphous Semiconductors: Structural, Optical, and Electronic PropertiesFrom EverandAmorphous Semiconductors: Structural, Optical, and Electronic PropertiesNo ratings yet
- Computational Methods in Lanthanide and Actinide ChemistryFrom EverandComputational Methods in Lanthanide and Actinide ChemistryMichael DolgNo ratings yet
- Settlement Analysis (Two Way Drainage)Document5 pagesSettlement Analysis (Two Way Drainage)Tanmoy DasNo ratings yet
- Pile Analysis, Design and DetailingDocument15 pagesPile Analysis, Design and DetailingMovies PointNo ratings yet
- DL2250,3000,4500,6000 PDFDocument138 pagesDL2250,3000,4500,6000 PDFPedro Pablo GonzalezNo ratings yet
- Procedure: Activity 2: Where in The World Is The Philippines? Part IiDocument28 pagesProcedure: Activity 2: Where in The World Is The Philippines? Part Iisamn cadNo ratings yet
- Radiation Protection & Quality Assurance in CT 2018Document51 pagesRadiation Protection & Quality Assurance in CT 2018abafzNo ratings yet
- W1 B Plane - Strain - TransformationDocument29 pagesW1 B Plane - Strain - TransformationSafwan RawiNo ratings yet
- Lesson+5+ +air Conditioning+and+Indoor+Air+QualityDocument53 pagesLesson+5+ +air Conditioning+and+Indoor+Air+QualityAmelia Isabelle VilagaNo ratings yet
- MCQ Chapter 2 ElectrochemistryDocument4 pagesMCQ Chapter 2 ElectrochemistryFact GamerNo ratings yet
- 5th Notes SST Revised FormatDocument5 pages5th Notes SST Revised FormatManzoor AlamNo ratings yet
- Dynamic Analysis of Pile Foundations Bem-Fem ModelDocument14 pagesDynamic Analysis of Pile Foundations Bem-Fem ModelShravan KumarNo ratings yet
- Product Group Industrial Vacuum Cleaners, 0415-156Document8 pagesProduct Group Industrial Vacuum Cleaners, 0415-156هبال حمزةNo ratings yet
- 2018-Effect of PVA On Mechanical Properties of Tapioca Starch Reinforced Coconut Fiber Biopolymer Composite PDFDocument5 pages2018-Effect of PVA On Mechanical Properties of Tapioca Starch Reinforced Coconut Fiber Biopolymer Composite PDFSlim ShaddysNo ratings yet
- Anchors in Cracked Concrete - Hilti SingaporeDocument16 pagesAnchors in Cracked Concrete - Hilti SingaporeOGStage2 ASNo ratings yet
- Thermodynamics 1655870560521Document26 pagesThermodynamics 1655870560521Singh DhruvNo ratings yet
- A Practical Guide To Soil-Structure Interaction: FEMA P-2091 / December 2020Document218 pagesA Practical Guide To Soil-Structure Interaction: FEMA P-2091 / December 2020Asrat Worku100% (1)
- Sample PT ProcedureDocument10 pagesSample PT ProcedureLarry Keating100% (2)
- Tool Life, Tool Wear Machinability PDFDocument9 pagesTool Life, Tool Wear Machinability PDFshivaNo ratings yet
- United States Patent (10) Patent No.: US 6,499,306 B2: Gillen (45) Date of Patent: Dec. 31, 2002Document25 pagesUnited States Patent (10) Patent No.: US 6,499,306 B2: Gillen (45) Date of Patent: Dec. 31, 2002HP ghnNo ratings yet
- Reversible Aqueous Zinc/manganese Oxide Energy Storage From Conversion ReactionsDocument7 pagesReversible Aqueous Zinc/manganese Oxide Energy Storage From Conversion ReactionsShofiNo ratings yet
- Caleffi: Maxcal Automatic Air VentDocument2 pagesCaleffi: Maxcal Automatic Air VentelmerbarrerasNo ratings yet
- Motions Class-IX (Physics) Solved Test Paper - 01Document4 pagesMotions Class-IX (Physics) Solved Test Paper - 01anshika tembhareNo ratings yet
- Type of Heat ExchangersDocument46 pagesType of Heat ExchangersguravdrNo ratings yet
- 7-Boiler - Eng - Ono SuparnoDocument14 pages7-Boiler - Eng - Ono SuparnoRaihan Rivellino AdzaniNo ratings yet
- H2 and CH4 Sorption On Cu-BTC Metal Organic Framew PDFDocument7 pagesH2 and CH4 Sorption On Cu-BTC Metal Organic Framew PDFMosllem ShahmohammadiNo ratings yet
- Aerial TriangulationDocument37 pagesAerial TriangulationRussel Clarete100% (1)
- Chapter 6 - Fatigue FailuresDocument70 pagesChapter 6 - Fatigue FailuresKanakNo ratings yet