Download as docx, pdf, or txt
You might also like
- OB Sample ReportsfdfadsDocument6 pagesOB Sample ReportsfdfadsJeffrey RamosNo ratings yet
- Sample POMRDocument4 pagesSample POMRJeffrey RamosNo ratings yet
- Systemic Pathology Laboratory ManualDocument58 pagesSystemic Pathology Laboratory ManualJeffrey RamosNo ratings yet
- Marcus 1997Document7 pagesMarcus 1997Araceli Enríquez OvandoNo ratings yet
- Cópia de Steinberg - Nihms-386599 - 2012Document22 pagesCópia de Steinberg - Nihms-386599 - 2012Marcio Lucio Candor MorillaNo ratings yet
- Blood Reviews Volume 26 Issue Supp-S1 2012 (Doi 10.1016/s0268-960x (12) 70010-3) Elliott Vichinsky - Advances in The Treatment of Alpha-ThalassemiaDocument4 pagesBlood Reviews Volume 26 Issue Supp-S1 2012 (Doi 10.1016/s0268-960x (12) 70010-3) Elliott Vichinsky - Advances in The Treatment of Alpha-ThalassemiaLink BuiNo ratings yet
- LP 10 Anemia 3Document43 pagesLP 10 Anemia 3Anonymous elq7jZiSNo ratings yet
- Hemoglobin Called Hemoglobin S or Hbs or Sickle Hemoglobin, in The Red Blood CellsDocument13 pagesHemoglobin Called Hemoglobin S or Hbs or Sickle Hemoglobin, in The Red Blood CellsPhương Ly LêNo ratings yet
- 2015 Biomédica Genetic Variants Associated With Fetal Hemoglobin Levels Show The Diverse Ethnic Origin....Document7 pages2015 Biomédica Genetic Variants Associated With Fetal Hemoglobin Levels Show The Diverse Ethnic Origin....Alejandro1-7No ratings yet
- 75 PDFDocument4 pages75 PDFJR VendeventerNo ratings yet
- Thalassemias: Alissa Martin,, Alexis A. ThompsonDocument9 pagesThalassemias: Alissa Martin,, Alexis A. ThompsonAmada RosalesNo ratings yet
- Case Report Non-infection Unit Β-Major ThalassemiaDocument27 pagesCase Report Non-infection Unit Β-Major ThalassemiaimamkdNo ratings yet
- Week 3: Hemoglobinopathies: HB Lepore Hbe Hbs HBC HB SC Disease HPFHDocument24 pagesWeek 3: Hemoglobinopathies: HB Lepore Hbe Hbs HBC HB SC Disease HPFHUdyani AgustinaNo ratings yet
- HEMA PracticalDocument14 pagesHEMA PracticalNanik AndianiNo ratings yet
- Review Article Role Of Xmni Polymorphism In Hbf Induction In Hbe/Β And Β-Thalassaemia PatientsDocument10 pagesReview Article Role Of Xmni Polymorphism In Hbf Induction In Hbe/Β And Β-Thalassaemia PatientsRumana Mahtarin TrishaNo ratings yet
- Review Article: A Review On Hemophilia in ChildrenDocument14 pagesReview Article: A Review On Hemophilia in ChildrenMelisa ClaireNo ratings yet
- HbE - THALASSEMIA A CASE REPORTDocument2 pagesHbE - THALASSEMIA A CASE REPORTHarshika KDGNo ratings yet
- Automated Capillary Electrophoresis in The ScreeningDocument9 pagesAutomated Capillary Electrophoresis in The Screeningsomething privateNo ratings yet
- HemoglobinopathiesDocument3 pagesHemoglobinopathiesMohamed Abdullahi OsmanNo ratings yet
- Hem 2020000102 CDocument7 pagesHem 2020000102 CEvan ElianNo ratings yet
- ThalassemiaDocument14 pagesThalassemiaHarshya RajeevanNo ratings yet
- Interpretation of Newborn Hemoglobin Screening Results Sep2013Document7 pagesInterpretation of Newborn Hemoglobin Screening Results Sep2013DR_Alaa_FakhriNo ratings yet
- HemoglobinDocument41 pagesHemoglobinSri Ram 07No ratings yet
- ThalassemiaDocument44 pagesThalassemiaitsshaswatNo ratings yet
- Erythrocyte Disorders in The Perinatal Period in AdversePregnancy Outcome and The Fetus:NeonateDocument12 pagesErythrocyte Disorders in The Perinatal Period in AdversePregnancy Outcome and The Fetus:Neonatethanhhien900No ratings yet
- ThalassemiaDocument93 pagesThalassemiaHafdzi MaulanaNo ratings yet
- ThalassaemiasDocument8 pagesThalassaemiasSalim UddinkhanNo ratings yet
- TestDocument2 pagesTestdvisnjaNo ratings yet
- ThalassaemiasDocument8 pagesThalassaemiasSalim UddinkhanNo ratings yet
- Hemoglobinopathies Hanna Waleed MohamedDocument10 pagesHemoglobinopathies Hanna Waleed Mohamedx8w6df2fxwNo ratings yet
- Introduction To Hemoglobinopathies Diagnostics On HPLC by P.C. GiordanoDocument4 pagesIntroduction To Hemoglobinopathies Diagnostics On HPLC by P.C. GiordanoUMMID WashimNo ratings yet
- HB Bart Fact SheetDocument2 pagesHB Bart Fact Sheetstrawberry pieNo ratings yet
- Sickle Cell Disease in PregnancyDocument18 pagesSickle Cell Disease in Pregnancyapi-370504667% (3)
- C 22Document8 pagesC 22Dharati PatelNo ratings yet
- 1303 Manuscript 5599 1 10 20170901Document8 pages1303 Manuscript 5599 1 10 20170901hariprem26No ratings yet
- 2-Perdida de La AHSP y Su Efecto en La Eritropoyesis y Beta-Talasemia JCI 2004Document10 pages2-Perdida de La AHSP y Su Efecto en La Eritropoyesis y Beta-Talasemia JCI 2004Mauri ParadedaNo ratings yet
- B-Talasemias. N Engl J Med 2021.Document17 pagesB-Talasemias. N Engl J Med 2021.VirginiaÁlvarezYepesNo ratings yet
- Sickle Cell DiseaseDocument18 pagesSickle Cell DiseasedrheayNo ratings yet
- Reactivation of Fetal Hemoglobin for Treating β-Thalassemia and Sickle Cell DiseaseDocument26 pagesReactivation of Fetal Hemoglobin for Treating β-Thalassemia and Sickle Cell Diseaseglory allexNo ratings yet
- лекции англDocument17 pagesлекции англBrett StevensonNo ratings yet
- Thalassemia Case ReportDocument40 pagesThalassemia Case ReportZarvi Jane MangubatNo ratings yet
- Talasemias RevisionDocument17 pagesTalasemias Revisionrodolfo venegasNo ratings yet
- 2 - HEMOGLOBIN HBCC, HbEEDocument10 pages2 - HEMOGLOBIN HBCC, HbEEAhmed AliNo ratings yet
- Alpha-Thalassemia: Authors: Renzo Galanello, MD Antonio Cao, MDDocument31 pagesAlpha-Thalassemia: Authors: Renzo Galanello, MD Antonio Cao, MDKiran KumarNo ratings yet
- Sickle Cell DiseaseDocument26 pagesSickle Cell DiseasebassamhematolNo ratings yet
- HemoglobinopathiesDocument26 pagesHemoglobinopathiesSanjey GaneshNo ratings yet
- Hemoglobinopathy MDDocument5 pagesHemoglobinopathy MDamaya rajivNo ratings yet
- Detection and Quantitation of Normal and Variant Haemoglobins: An Analytical ReviewDocument15 pagesDetection and Quantitation of Normal and Variant Haemoglobins: An Analytical ReviewAmanda MatthewsNo ratings yet
- Abid 3 PDFDocument14 pagesAbid 3 PDFLuan MatosNo ratings yet
- Abid 3 PDFDocument14 pagesAbid 3 PDFLuan MatosNo ratings yet
- Hemoglobin C DiseaseDocument14 pagesHemoglobin C DiseaseKashan SiddiquiNo ratings yet
- Sickle Cell Diseases and Thalasemia Asma2Document60 pagesSickle Cell Diseases and Thalasemia Asma2Female calmNo ratings yet
- Hemoglobinopathies and ThalassemiasDocument10 pagesHemoglobinopathies and ThalassemiashartNo ratings yet
- HbE and Beta Thalassemia and Oxidative StressDocument20 pagesHbE and Beta Thalassemia and Oxidative StressRaehana AlaydrusNo ratings yet
- Chapter 28 Summary ThalassemiaDocument10 pagesChapter 28 Summary ThalassemiasanastrikepoNo ratings yet
- Thalasemia MedscapeDocument21 pagesThalasemia MedscapeFenty IswarNo ratings yet
- 3-T and Sicke Cell Disease 2016Document68 pages3-T and Sicke Cell Disease 2016ThaveeshaLindsayWhiteNo ratings yet
- HemoglobinDocument7 pagesHemoglobinNinik Triayu S100% (2)
- ThalassemiaDocument3 pagesThalassemiaNajeebullah MujahidNo ratings yet
- HaemoglobinopathiesDocument7 pagesHaemoglobinopathiesAdham TarekNo ratings yet
- A Model for Gene Therapy: Gene Replacement in the Treatment of Sickle Cell Anemia and ThalassemiaFrom EverandA Model for Gene Therapy: Gene Replacement in the Treatment of Sickle Cell Anemia and ThalassemiaNo ratings yet
- IDS TablesDocument2 pagesIDS TablesJeffrey RamosNo ratings yet
- Geriatric PHSDFDSFDocument4 pagesGeriatric PHSDFDSFJeffrey RamosNo ratings yet
- Digestive System Cont PDFDocument10 pagesDigestive System Cont PDFJeffrey RamosNo ratings yet
- Issues On HIV and STI Among Pregnant Women: DR Helen Madamba, MD MPH-TM Fpogs FpidsogDocument32 pagesIssues On HIV and STI Among Pregnant Women: DR Helen Madamba, MD MPH-TM Fpogs FpidsogJeffrey RamosNo ratings yet
- 406-01. Manuscript (Article Text) - 1308-1-10-20130405Document4 pages406-01. Manuscript (Article Text) - 1308-1-10-20130405Jeffrey RamosNo ratings yet
- Legal Med.: Archaeological Methods: Search and Recovery/Survey and ExcavationDocument2 pagesLegal Med.: Archaeological Methods: Search and Recovery/Survey and ExcavationJeffrey RamosNo ratings yet
- Alcohol Intake: Increased Serum Amylase and Serum LipaseDocument6 pagesAlcohol Intake: Increased Serum Amylase and Serum LipaseJeffrey RamosNo ratings yet
- RADIO Liver and GallbladderDocument6 pagesRADIO Liver and GallbladderJeffrey RamosNo ratings yet
- CD Rom 7 Slides Westgard Multirule SystemDocument11 pagesCD Rom 7 Slides Westgard Multirule SystemJeffrey RamosNo ratings yet
- FsfsfseeesxxvvbhklopgfDocument49 pagesFsfsfseeesxxvvbhklopgfJeffrey RamosNo ratings yet
- Anatomy PBL Lower Extremities 1Document2 pagesAnatomy PBL Lower Extremities 1Jeffrey RamosNo ratings yet
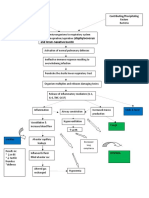
- Pathophysiology of PneumoniaDocument1 pagePathophysiology of PneumoniaJeffrey RamosNo ratings yet
- NM, / 'PDocument93 pagesNM, / 'PJeffrey RamosNo ratings yet
- Pathophysiology of PneumoniaDocument2 pagesPathophysiology of PneumoniaJeffrey Ramos100% (1)
- Sample POMRDocument4 pagesSample POMRJeffrey RamosNo ratings yet