Download as pdf or txt
You might also like
- Yann Tiersen EUSA PDFDocument57 pagesYann Tiersen EUSA PDFDaniela Greere100% (1)
- DP Lokwani - The ABC of CBC Interpretation of Complete Blood Count & Histograms 2EDocument1 pageDP Lokwani - The ABC of CBC Interpretation of Complete Blood Count & Histograms 2EWeal AlhaidaryNo ratings yet
- Haematology NotesDocument184 pagesHaematology NotesJason roy100% (1)
- Changes Include Addition of Nattokinase As First Line Therapy and Reordering of First-And Second-Line TherapiesDocument60 pagesChanges Include Addition of Nattokinase As First Line Therapy and Reordering of First-And Second-Line TherapiesJoshua VanhieNo ratings yet
- Term 3 Rationale Pharmacology and MCNDocument35 pagesTerm 3 Rationale Pharmacology and MCNKing KongNo ratings yet
- WBC BasicsDocument70 pagesWBC BasicsZoe ZillaNo ratings yet
- Orac MethodDocument5 pagesOrac MethodPriscillaL.SilvaNo ratings yet
- English For Waiters BCDocument85 pagesEnglish For Waiters BCelenbaran100% (11)
- Merck Vitamin K Package Insert - Aquamephyton PIDocument5 pagesMerck Vitamin K Package Insert - Aquamephyton PIDonna100% (10)
- Bloody Easy 4Document82 pagesBloody Easy 4dubblewalker100% (1)
- Alkaline Phpsphatase Blt00003 4Document2 pagesAlkaline Phpsphatase Blt00003 4Erick AlvarezNo ratings yet
- Predanalitika KoagulacijaDocument10 pagesPredanalitika KoagulacijaAnonymous w4qodCJNo ratings yet
- Alpha Fetoprotein (Afp)Document10 pagesAlpha Fetoprotein (Afp)Andi UkengNo ratings yet
- The Diagnostic Use of ADVIA 2120i Siemens and An "APL Criteria" CanDocument9 pagesThe Diagnostic Use of ADVIA 2120i Siemens and An "APL Criteria" CananggaririnNo ratings yet
- Comprehensive Report On Rapid Plasma Reagin Test (RPR)Document3 pagesComprehensive Report On Rapid Plasma Reagin Test (RPR)Kim RuizNo ratings yet
- Cast in Urine SedimentDocument3 pagesCast in Urine Sedimentfirie100% (1)
- In Vitro MarketDocument59 pagesIn Vitro MarketmastbittuNo ratings yet
- HPLC Studies in HemoglobinopathiesDocument6 pagesHPLC Studies in HemoglobinopathiesDevi SusantiNo ratings yet
- Slide Preparation of Cerebrospinal Fluid For Cytological ExaminationDocument3 pagesSlide Preparation of Cerebrospinal Fluid For Cytological ExaminationMurshed HaidarNo ratings yet
- Routine Laboratory Evaluation of CoagulationDocument7 pagesRoutine Laboratory Evaluation of CoagulationGilo IlaganNo ratings yet
- Kaplan: Clinical Chemistry, 5 Edition: Clinical References - Methods of AnalysisDocument9 pagesKaplan: Clinical Chemistry, 5 Edition: Clinical References - Methods of AnalysispudjoNo ratings yet
- Clinical Manifestations and Diagnosis of The Thalassemias - UpToDateDocument52 pagesClinical Manifestations and Diagnosis of The Thalassemias - UpToDatesushi37No ratings yet
- Blood ComponentsDocument44 pagesBlood ComponentsKrisha VittoNo ratings yet
- Lab 2 ImmunohaematologyDocument3 pagesLab 2 ImmunohaematologyLorraine ThompsonNo ratings yet
- Chemical Pathology HODocument17 pagesChemical Pathology HOMurali TiarasanNo ratings yet
- TSH Acculite Clia Rev 4Document2 pagesTSH Acculite Clia Rev 4ghumantuNo ratings yet
- 300-5208 B BFM Clsi SopDocument23 pages300-5208 B BFM Clsi SopnjujjnjnjjnnjNo ratings yet
- Transfusion Training Checklist May 2015Document5 pagesTransfusion Training Checklist May 2015dheNo ratings yet
- Automated Versus Manual Platelet Count in Aden 2161 0681-3-149Document4 pagesAutomated Versus Manual Platelet Count in Aden 2161 0681-3-149Jeffry MaglalangNo ratings yet
- Enzymes - PPT 1Document54 pagesEnzymes - PPT 1Cesar Augusto Airampo Macedo100% (1)
- Techtalk August2010Document2 pagesTechtalk August2010Abu KhalidNo ratings yet
- Biochemistry AnalyzerDocument23 pagesBiochemistry AnalyzerBunga ExsotikaNo ratings yet
- 2019 Modified Blood ComponentsDocument30 pages2019 Modified Blood Componentsسعد الطائعNo ratings yet
- Glucose Arc ChemDocument8 pagesGlucose Arc ChemDrFarah Emad AliNo ratings yet
- 10 Lupus Anticoagulants 1Document28 pages10 Lupus Anticoagulants 1medico medicoNo ratings yet
- Alpha FetoproteinDocument2 pagesAlpha FetoproteinmonimoyNo ratings yet
- Principles, Applications and InterpretationsDocument64 pagesPrinciples, Applications and InterpretationsAbbi Yanto ArtNo ratings yet
- Lab HemostasisDocument29 pagesLab HemostasisTingLi Lucia Lorigiano100% (2)
- Uric Acid Mono SL: Clinical SignificanceDocument2 pagesUric Acid Mono SL: Clinical SignificancexlkoNo ratings yet
- Alloimmunisation To Blood Group AntigensDocument34 pagesAlloimmunisation To Blood Group AntigensbloodbankNo ratings yet
- Analyte Stability & Freeze-Thaw Information-1Document8 pagesAnalyte Stability & Freeze-Thaw Information-1Yusuf Indra SentosaNo ratings yet
- Red Blood Cell DisordersDocument106 pagesRed Blood Cell DisordersCharmaine Clarke-BlytheNo ratings yet
- 11013003Document1 page11013003Johnmar AquinoNo ratings yet
- Written Report in Analysis of Urine and Other Body Fluids: (Cerebrospinal Fluid)Document12 pagesWritten Report in Analysis of Urine and Other Body Fluids: (Cerebrospinal Fluid)Janielle FajardoNo ratings yet
- IFCC TF Ethics in Lab MedicineDocument16 pagesIFCC TF Ethics in Lab MedicineDr.Deepanshu SinghalNo ratings yet
- Acute Promyelocytic LeukemiaDocument46 pagesAcute Promyelocytic LeukemiaKartthik ShanmugamNo ratings yet
- 2.5 Antibody ScreeningDocument5 pages2.5 Antibody ScreeningBALAJINo ratings yet
- Vdoc - Pub Diagnostic Cytopathology Expert Consult Online and PrintDocument947 pagesVdoc - Pub Diagnostic Cytopathology Expert Consult Online and PrintAreesha ArifNo ratings yet
- The Significance of The Hemoglobin A2Document18 pagesThe Significance of The Hemoglobin A2Paula MadureuraNo ratings yet
- Blood Component PreparationDocument24 pagesBlood Component PreparationLaiba ArshadNo ratings yet
- Immuno HematologyDocument24 pagesImmuno HematologyHoraa H AL-marhoonNo ratings yet
- Haematologists Toolkit V1b August 16 EoEDocument23 pagesHaematologists Toolkit V1b August 16 EoELlrss AdnNo ratings yet
- Compatibility Testing: Week 5Document33 pagesCompatibility Testing: Week 5Bridgette100% (1)
- Radial ImmunoDocument8 pagesRadial Immunograce meunierNo ratings yet
- Plasma Proteins in Disease DiagnosisDocument81 pagesPlasma Proteins in Disease DiagnosisSaaqo Qasim100% (1)
- Transfusion Practice Guidelines For Clinic and Laboratory PersonnelDocument98 pagesTransfusion Practice Guidelines For Clinic and Laboratory PersonnelZhayreal Zaki100% (1)
- ROTEM AnalysisDocument42 pagesROTEM AnalysisAnnie ArriNo ratings yet
- Routine Laboratory Evaluation of CoagulationDocument32 pagesRoutine Laboratory Evaluation of CoagulationArshie08No ratings yet
- Packed Cell Volume (PCV) TestDocument3 pagesPacked Cell Volume (PCV) TestchineduNo ratings yet
- Blood Grouping ReagentsDocument7 pagesBlood Grouping ReagentsDominic EmerencianaNo ratings yet
- Pocket Guide On Red Cells 2012Document8 pagesPocket Guide On Red Cells 2012Dave OrlandoNo ratings yet
- Laboratory Tests For EndocrinologyDocument6 pagesLaboratory Tests For EndocrinologyAnastasia100% (1)
- Enseval Examination of Bacterial Contamination in Blood Components Biomerieux PDFDocument21 pagesEnseval Examination of Bacterial Contamination in Blood Components Biomerieux PDFCik KahadiNo ratings yet
- Blood Bank 4 DiscpDocument20 pagesBlood Bank 4 DiscpHector de la CruzNo ratings yet
- Endocrinology Guide TransgenderDocument35 pagesEndocrinology Guide TransgenderDaniela Greere0% (1)
- Toxicologie - Notiuni IntroductiveDocument15 pagesToxicologie - Notiuni IntroductiveDaniela GreereNo ratings yet
- Subiecte Engleza BilingvDocument1 pageSubiecte Engleza BilingvDaniela GreereNo ratings yet
- Bilingv - Subiecte OralDocument1 pageBilingv - Subiecte OralDaniela GreereNo ratings yet
- Guide To Sun Danese MusicDocument117 pagesGuide To Sun Danese MusicDaniela Greere100% (1)
- Acute Deep Venous ThrombosisDocument92 pagesAcute Deep Venous ThrombosisDeepak Kumar100% (2)
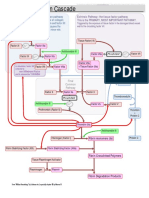
- Coagulation CascadeDocument5 pagesCoagulation Cascadepieterinpretoria391100% (1)
- Blood: Jeff Tjong, PHDDocument59 pagesBlood: Jeff Tjong, PHDyat yat szeNo ratings yet
- Hemostasis Practice: State-Of-The-Art: Giuseppe Lippi, Emmanuel J. FavaloroDocument6 pagesHemostasis Practice: State-Of-The-Art: Giuseppe Lippi, Emmanuel J. FavaloroRilia IrianiNo ratings yet
- Papel Inmunologico de Hookworm. 21th Century. Brooker 2004.Document59 pagesPapel Inmunologico de Hookworm. 21th Century. Brooker 2004.Karen JulianaNo ratings yet
- Hemoststic AgentsDocument17 pagesHemoststic AgentsJoseph John K PothanikatNo ratings yet
- Coagulation Made EasyDocument27 pagesCoagulation Made EasyHaris Tan100% (1)
- HaemophiliaDocument15 pagesHaemophiliaSanmuga VimalanathanNo ratings yet
- Administration of Coagulation-Altering Therapy in The Patient Presenting For Oral Health and Maxillofacial SurgeryDocument18 pagesAdministration of Coagulation-Altering Therapy in The Patient Presenting For Oral Health and Maxillofacial SurgeryLaura Giraldo QuinteroNo ratings yet
- Hemostasis, Clotting Disorder and AnticoagulantsDocument87 pagesHemostasis, Clotting Disorder and AnticoagulantsSomit Jain100% (1)
- Anticoagulant Activities of Curcumin and Its DerivativeDocument6 pagesAnticoagulant Activities of Curcumin and Its DerivativeAlfred YangaoNo ratings yet
- Summary of Product CharacteristicsDocument24 pagesSummary of Product Characteristicsddandan_2No ratings yet
- Platelets. Hemostasis.: Learning ObjectivesDocument37 pagesPlatelets. Hemostasis.: Learning ObjectivesQasim alaliNo ratings yet
- Blood Physiology MainDocument68 pagesBlood Physiology MainOmenaalaNo ratings yet
- Anticoagulants DaniellewelschmeyerDocument30 pagesAnticoagulants Daniellewelschmeyersumanth gowdaNo ratings yet
- 7th Edition 7.1Document50 pages7th Edition 7.1eshwar_org100% (1)
- Hemostasis and Fibrinolysis: Adelina VladDocument82 pagesHemostasis and Fibrinolysis: Adelina VladLoly SinagaNo ratings yet
- Coag Made EasyDocument15 pagesCoag Made Easygus_lionsNo ratings yet
- Overcoming Protein Instability Problems During Fusion Protein CleavageDocument5 pagesOvercoming Protein Instability Problems During Fusion Protein CleavageAsmaNo ratings yet
- Bloody Easy - Coagulation ExplainedDocument25 pagesBloody Easy - Coagulation ExplainedDaniela GreereNo ratings yet
- Physio 2.05 Bloodphysiology2 HemostasisDocument9 pagesPhysio 2.05 Bloodphysiology2 HemostasisSimon Peter Familara100% (1)
- Clsi Poct14-2020Document68 pagesClsi Poct14-2020RAHAOUI MehdiNo ratings yet
- Coagulation Factors: An OverviewDocument13 pagesCoagulation Factors: An OverviewMonnierNo ratings yet
- Practical Obstetric Hematology PDFDocument208 pagesPractical Obstetric Hematology PDFSteve CullenNo ratings yet
- Survey of Ethno Medicinal Plant Showing Wound Healing ActivityDocument18 pagesSurvey of Ethno Medicinal Plant Showing Wound Healing ActivityIJAR JOURNALNo ratings yet