Download as pdf or txt
You might also like
- 2 Theory of Errors in ObservationsDocument33 pages2 Theory of Errors in ObservationsJerick Cosiñero0% (1)
- Johnson Cook Fortran CodeDocument90 pagesJohnson Cook Fortran CodeJoe SatchNo ratings yet
- Ngss and CSTDocument8 pagesNgss and CSTapi-261602385No ratings yet
- Thermophysical Properties of Fluids Dynamic Viscos PDFDocument13 pagesThermophysical Properties of Fluids Dynamic Viscos PDFOmodolor StevedanNo ratings yet
- An Introduction To Molecular ModelingDocument10 pagesAn Introduction To Molecular ModelingVidya MNo ratings yet
- Proof of Concept For Fast Equation of State Development Using An Integrated Experimental-Computational ApproachDocument20 pagesProof of Concept For Fast Equation of State Development Using An Integrated Experimental-Computational ApproachMustapha MaaroufNo ratings yet
- Electronic Descriptors For Supervised SpectroscopiDocument9 pagesElectronic Descriptors For Supervised SpectroscopianaNo ratings yet
- On The Applicability of The Uniquac Method To Ternary Liquid - Liquid EquilibriaDocument10 pagesOn The Applicability of The Uniquac Method To Ternary Liquid - Liquid EquilibriaHero19No ratings yet
- Lightcraft PropulsionDocument37 pagesLightcraft PropulsionLin CornelisonNo ratings yet
- Systematic Validation and Improvement of Quantum Chemistry Methods For The Prediction of Physical and Chemical PropertiesDocument2 pagesSystematic Validation and Improvement of Quantum Chemistry Methods For The Prediction of Physical and Chemical PropertiesElectro_LiteNo ratings yet
- Heat and ChargeDocument18 pagesHeat and ChargeBhanu PrakashNo ratings yet
- Water Saft-Vr MieDocument32 pagesWater Saft-Vr MiePatrice PariNo ratings yet
- Discussion On Criterion of Determination of The Kinetic Parameters of The Linear Heating ReactionsDocument13 pagesDiscussion On Criterion of Determination of The Kinetic Parameters of The Linear Heating ReactionsdandanNo ratings yet
- Application of The Method of Characteristics in The Analysis of Transient Events in Natural Gas Distribution NetworksDocument8 pagesApplication of The Method of Characteristics in The Analysis of Transient Events in Natural Gas Distribution NetworksIJAERS JOURNALNo ratings yet
- Rths With Concurrent Model Updating On A Distributed PlatformDocument7 pagesRths With Concurrent Model Updating On A Distributed PlatformfradmardNo ratings yet
- Quantum Chemical Descriptors in QSAR QSPR StudiesDocument17 pagesQuantum Chemical Descriptors in QSAR QSPR StudiesCarlos Alberto Bayona LópezNo ratings yet
- Study of The Thermal Behaviour of PVC in The Presence of Zeolites and A SepiloiteDocument1 pageStudy of The Thermal Behaviour of PVC in The Presence of Zeolites and A SepiloiteSteven ObrienNo ratings yet
- Estimating The Electrical Conductivity of Human Tissue in Radiofrequency Hyperthermia TherapyDocument10 pagesEstimating The Electrical Conductivity of Human Tissue in Radiofrequency Hyperthermia Therapyjesusortega2961No ratings yet
- Predicting The Hydrate FormationDocument10 pagesPredicting The Hydrate FormationmviteazuNo ratings yet
- Mulero Parra 08 1Document16 pagesMulero Parra 08 1Mustapha MaaroufNo ratings yet
- Bi Stability and Oscillations in Chemical Reaction NetworksDocument35 pagesBi Stability and Oscillations in Chemical Reaction Networksbwu008No ratings yet
- Rsuq 2016 002Document39 pagesRsuq 2016 002Vinícius Ribeiro MachadoNo ratings yet
- 2007 Loukas BlendsDocument11 pages2007 Loukas BlendsJavier Ramos SpotifyNo ratings yet
- Lattice 2008 099Document7 pagesLattice 2008 099Red GluonNo ratings yet
- Predictive Group Contribution Models For The Thermophysical Properties of Ionic LiquidsDocument17 pagesPredictive Group Contribution Models For The Thermophysical Properties of Ionic LiquidsMohammad_Ismai_3096No ratings yet
- XX Paper 24Document9 pagesXX Paper 24VindhyaNo ratings yet
- Xavier Rozanska, James J. P. Stewart, Philippe Ungerer, Benoit Leblanc, Clive Freeman, Paul Saxe, and Erich WimmerDocument8 pagesXavier Rozanska, James J. P. Stewart, Philippe Ungerer, Benoit Leblanc, Clive Freeman, Paul Saxe, and Erich WimmerDaniel DominguezNo ratings yet
- Magneto HydrodynamicsDocument32 pagesMagneto HydrodynamicsNitin PooniaNo ratings yet
- Yaroshchyk 2006Document10 pagesYaroshchyk 2006enzopcontreras044No ratings yet
- ResbDocument10 pagesResbRia Sinha RoyNo ratings yet
- A New Approach For Optimizing A Bipolar Charge Transport Model For Dielectric Materials Theoretical Framework PDFDocument8 pagesA New Approach For Optimizing A Bipolar Charge Transport Model For Dielectric Materials Theoretical Framework PDFAmrendra kumarNo ratings yet
- Modelling in Aquatic Chemistry 2Document776 pagesModelling in Aquatic Chemistry 2paul mulwaNo ratings yet
- Scienti C Editors/ Editeurs Scienti Ques Contributors/CollaborateursDocument12 pagesScienti C Editors/ Editeurs Scienti Ques Contributors/CollaborateursthefunfundaNo ratings yet
- Bond Graph Modelling For Chemical ReactorsDocument17 pagesBond Graph Modelling For Chemical ReactorsMejbahul SarkerNo ratings yet
- JafariDocument18 pagesJafariCeleron DNo ratings yet
- Multi-Scale Modeling in Materials Science and EngineeringDocument12 pagesMulti-Scale Modeling in Materials Science and EngineeringonebyzerooutlookNo ratings yet
- MATI612024pp15 38Document26 pagesMATI612024pp15 38sneha.921530sharmaNo ratings yet
- TGDocument6 pagesTGYeferson GuevaraNo ratings yet
- Designs: Analytical Expression of Parabolic Trough Solar Collector PerformanceDocument17 pagesDesigns: Analytical Expression of Parabolic Trough Solar Collector PerformanceArunsinghNo ratings yet
- Correlation Between The Glass Transition Temperatures and Repeating Unit Structure For High Molecular Weight PolymersDocument8 pagesCorrelation Between The Glass Transition Temperatures and Repeating Unit Structure For High Molecular Weight PolymersSinisa Gale GacicNo ratings yet
- Subject Physics: PAPER No. 10: Statistical Mechanics Module No7.Document15 pagesSubject Physics: PAPER No. 10: Statistical Mechanics Module No7.Srinivasu RaoNo ratings yet
- Data Analytics Approach To Predict High-TemperatureDocument33 pagesData Analytics Approach To Predict High-Temperaturebmalki68No ratings yet
- Lecture-3 - Role of Chemistry in Computer ScienceDocument11 pagesLecture-3 - Role of Chemistry in Computer Sciencemdeaqub hasanNo ratings yet
- Calculation of Critical Temperatures by Empirical FormulaeDocument6 pagesCalculation of Critical Temperatures by Empirical FormulaeAndress SsalomonnNo ratings yet
- Lifetime Prediction of Thermo-Mechanical Fatigue For Exhaust ManifoldDocument10 pagesLifetime Prediction of Thermo-Mechanical Fatigue For Exhaust Manifoldram shyamNo ratings yet
- Mechanics of Materials: Kamran A. Khan, Romina Barello, Anastasia H. Muliana, Martin LévesqueDocument18 pagesMechanics of Materials: Kamran A. Khan, Romina Barello, Anastasia H. Muliana, Martin LévesqueLuthfi AdyNo ratings yet
- DTMM and COSMIC Molecular Mechanics Parameters For AlkylsilanesDocument14 pagesDTMM and COSMIC Molecular Mechanics Parameters For AlkylsilanesLuu Xuan CuongNo ratings yet
- Quantum Chaos in The Nuclear Collective Model: I. Classical-Quantum CorrespondenceDocument10 pagesQuantum Chaos in The Nuclear Collective Model: I. Classical-Quantum CorrespondenceBayer MitrovicNo ratings yet
- Nicol 2001Document19 pagesNicol 2001Clarence AG YueNo ratings yet
- Spectroscopic and Theoretical AspectsDocument123 pagesSpectroscopic and Theoretical AspectsMPCNo ratings yet
- Binary Interaction ParametersDocument16 pagesBinary Interaction ParametersSchöberl ErichNo ratings yet
- Bond Dissociation Energies For Diatomic Molecules Containing 3d Transition Metals: Benchmark Scalar Relativistic Coupled Cluster Calculations For Twenty MoleculesDocument40 pagesBond Dissociation Energies For Diatomic Molecules Containing 3d Transition Metals: Benchmark Scalar Relativistic Coupled Cluster Calculations For Twenty MoleculesNyau NyauNo ratings yet
- Text Kuliah Metoda Statistika QSAR KIMED 2023 PDFDocument22 pagesText Kuliah Metoda Statistika QSAR KIMED 2023 PDFameljpNo ratings yet
- 2004FPENannoolalRareyRamjugernath 1 BoilingTemperatureEstimation PDFDocument19 pages2004FPENannoolalRareyRamjugernath 1 BoilingTemperatureEstimation PDFAriel MorónNo ratings yet
- Geosciences: Data-Driven Geothermal Reservoir Modeling: Estimating Permeability Distributions by Machine LearningDocument18 pagesGeosciences: Data-Driven Geothermal Reservoir Modeling: Estimating Permeability Distributions by Machine LearningYounes ZizouNo ratings yet
- Livingstone 2001Document12 pagesLivingstone 2001sarah.echcherifNo ratings yet
- Hartree Fock Geometry Optimisation of Periodic Systems With The C CodeDocument8 pagesHartree Fock Geometry Optimisation of Periodic Systems With The C CodeLorena Monterrosas PérezNo ratings yet
- Finite Deformation Continuum Model For Single-Walled Carbon NanotubesDocument9 pagesFinite Deformation Continuum Model For Single-Walled Carbon NanotubesEduardo GarciaNo ratings yet
- JHM Lotuses Paper4Document12 pagesJHM Lotuses Paper4Muthu RajanNo ratings yet
- Multiple Translational Temperature Model and Its SDocument27 pagesMultiple Translational Temperature Model and Its SvaiNo ratings yet
- C6+ Characterization For Natural Gas - Chemical Process Engineering - Eng-TipsDocument3 pagesC6+ Characterization For Natural Gas - Chemical Process Engineering - Eng-Tipsknight282009No ratings yet
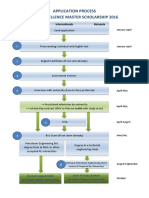
- OMV Scholarship Application ProcessDocument1 pageOMV Scholarship Application Processknight282009No ratings yet
- Form Membership GeneralDocument2 pagesForm Membership Generalknight282009100% (1)
- Hydrocarbon Dew Point SpecificationDocument17 pagesHydrocarbon Dew Point Specificationargentino_ar01No ratings yet
- 1 Notes On The Poisson Process: 1.1 Point ProcessesDocument19 pages1 Notes On The Poisson Process: 1.1 Point ProcessesKaran ShettyNo ratings yet
- Top QuestionsDocument8 pagesTop QuestionsgigiNo ratings yet
- Corning SMF28e XB Bend Optimized Fiber Spec SheetDocument2 pagesCorning SMF28e XB Bend Optimized Fiber Spec SheetcolinyNo ratings yet
- Nature News FeatureDocument8 pagesNature News FeaturebookreadermanNo ratings yet
- Training Servo Hydraulic Drive Systems 36Document61 pagesTraining Servo Hydraulic Drive Systems 36Gary Jenkins100% (4)
- Modern Physics Type 2 PART 2 of 3 ENGDocument21 pagesModern Physics Type 2 PART 2 of 3 ENGManav AgrawalNo ratings yet
- 21 Electrostatics-E FieldDocument5 pages21 Electrostatics-E FieldeltytanNo ratings yet
- Design of Liquid-Liquid Extraction ColumnsDocument36 pagesDesign of Liquid-Liquid Extraction ColumnsJowel MercadoNo ratings yet
- Cold Shrink Termination MVTI - MVTODocument6 pagesCold Shrink Termination MVTI - MVTOdes1982No ratings yet
- Algebra: Revision Sheet - Fp1 (Mei)Document10 pagesAlgebra: Revision Sheet - Fp1 (Mei)GadisNovelNo ratings yet
- Hydrometer - Fluids Pressure Upthrust and Flotation - TutorvistaDocument6 pagesHydrometer - Fluids Pressure Upthrust and Flotation - TutorvistaRamaKrishnanGNo ratings yet
- Quality Control Manual v2Document66 pagesQuality Control Manual v2Oladunni AfolabiNo ratings yet
- 3-D Bracket: Workshop 10A Loading and SolutionDocument26 pages3-D Bracket: Workshop 10A Loading and SolutionSathi MechNo ratings yet
- Introduction To Mobile Robotics - Burgard PDFDocument745 pagesIntroduction To Mobile Robotics - Burgard PDFSantiago Garrido BullónNo ratings yet
- Test SuccessDocument36 pagesTest SuccessAlPrince007No ratings yet
- Overload Clutches in Agricultural Machinery: Nienhaus, ClemensDocument6 pagesOverload Clutches in Agricultural Machinery: Nienhaus, ClemensgangatilfrettarNo ratings yet
- Zonal Cavity Method For Calculating The Number of Lamps in An AreaDocument8 pagesZonal Cavity Method For Calculating The Number of Lamps in An AreaAbdulyekini Ahmadu100% (1)
- 006 FrictionDocument19 pages006 FrictionJorge VaccarelloNo ratings yet
- SPT English PDFDocument4 pagesSPT English PDFPatolinoNo ratings yet
- Pacholski Practical MAM 6 2015Document4 pagesPacholski Practical MAM 6 2015dacaNo ratings yet
- SSA 3 TrebuchetDocument9 pagesSSA 3 TrebuchetemomNo ratings yet
- FRCR US Lecture 1Document76 pagesFRCR US Lecture 1TunnelssNo ratings yet
- Multipath FadingDocument18 pagesMultipath FadingJulia Joseph100% (1)
- PLC Based Automatic Liquid Filling System For Different Sized BottlesDocument6 pagesPLC Based Automatic Liquid Filling System For Different Sized BottlesRodel Gamale100% (1)
- Air Permeability and Costructional Parameters of Woven FabricsDocument6 pagesAir Permeability and Costructional Parameters of Woven FabricsDorin VladNo ratings yet
- Riemann Sums and The Definite Integral: Lesson 5.3Document17 pagesRiemann Sums and The Definite Integral: Lesson 5.3LEANo ratings yet
- Belt AnalysisDocument4 pagesBelt AnalysisMaheswaran MuthaiyanNo ratings yet
- Y 18 Hand Book - M.Tech - Y18 R (SE, CTM, GI) PDFDocument61 pagesY 18 Hand Book - M.Tech - Y18 R (SE, CTM, GI) PDFChandrakanth K JOeNo ratings yet
- Mod 6 Materials and Hardware: Exam PointsDocument16 pagesMod 6 Materials and Hardware: Exam PointsKetanRaoNo ratings yet