Download as pdf or txt
You might also like
- Exercises PolsDocument8 pagesExercises PolsAshutosh PandeyNo ratings yet
- StatisticalPhysics Part1 HandoutDocument27 pagesStatisticalPhysics Part1 HandoutMauro LaraNo ratings yet
- Chlorine Recovery From Hydrogen ChlorideDocument5 pagesChlorine Recovery From Hydrogen ChlorideAnne Porter100% (1)
- Thermal Physics Lecture 13Document7 pagesThermal Physics Lecture 13OmegaUserNo ratings yet
- Heat Capacity of An Ideal Polyatomic GasDocument11 pagesHeat Capacity of An Ideal Polyatomic GasRhaditNo ratings yet
- Thermal Physics Lecture 12Document8 pagesThermal Physics Lecture 12OmegaUserNo ratings yet
- 1.1 Principle of Statistical Physics and EnsemblesDocument9 pages1.1 Principle of Statistical Physics and EnsemblesArkayan LahaNo ratings yet
- Freely-Jointed Chain: For Up To Date Version of This Document, See Z. SuoDocument6 pagesFreely-Jointed Chain: For Up To Date Version of This Document, See Z. SuokhayatNo ratings yet
- Entropy. Temperature. Chemical Potential. Thermodynamic Identities. Third LawDocument20 pagesEntropy. Temperature. Chemical Potential. Thermodynamic Identities. Third LawAndam PluffNo ratings yet
- Momentum: Nirmaan Shanker October 2015Document7 pagesMomentum: Nirmaan Shanker October 2015Nirmaan ShankerNo ratings yet
- 1.1 Principle of Statistical Physics and EnsemblesDocument10 pages1.1 Principle of Statistical Physics and EnsemblesManu SharmaNo ratings yet
- Advanced Particle Physics - Lecture 1 Overview, Units and Notation 1Document3 pagesAdvanced Particle Physics - Lecture 1 Overview, Units and Notation 1epidermNo ratings yet
- Vibrations Phonons3Document53 pagesVibrations Phonons3Kartik Dutta67% (3)
- Ans 1Document12 pagesAns 1euphysics2025No ratings yet
- Stat ThermoDocument14 pagesStat ThermoskluxNo ratings yet
- PHY357 Lecture6Document23 pagesPHY357 Lecture6Sijil SalimNo ratings yet
- Gravitation: DZ e DZ e DZ Z PDocument4 pagesGravitation: DZ e DZ e DZ Z Pita lailaNo ratings yet
- UBC Condensed Matter 502 Notes Part1Document18 pagesUBC Condensed Matter 502 Notes Part1kahoNo ratings yet
- Equations of State For The Fermi Gas Model and A Realistic Model: Properties of Neutron StarsDocument24 pagesEquations of State For The Fermi Gas Model and A Realistic Model: Properties of Neutron StarsAbhass KumarNo ratings yet
- Degenrate Fermi GasDocument15 pagesDegenrate Fermi GasArjun IyerNo ratings yet
- Indistinguishable ParticlesDocument4 pagesIndistinguishable Particlesfwegfw3No ratings yet
- 7-Ensembles 0Document20 pages7-Ensembles 0loop arrayNo ratings yet
- School of Physics and Astronomy Junior Honours Thermodynamics GJA 2013-2014Document4 pagesSchool of Physics and Astronomy Junior Honours Thermodynamics GJA 2013-2014Francisco De León-Sotelo EstebanNo ratings yet
- Preparation For Midterm ExaminationDocument6 pagesPreparation For Midterm ExaminationĐức PhanNo ratings yet
- Rich ThinkingDocument17 pagesRich ThinkingMain AccountNo ratings yet
- Thermal Physics Lecture 24Document6 pagesThermal Physics Lecture 24OmegaUserNo ratings yet
- PartitionDocument2 pagesPartitionAshuNo ratings yet
- Compton ScatteringDocument6 pagesCompton Scatteringffr.gimenezNo ratings yet
- 3 Dimensional String TheoryDocument6 pages3 Dimensional String TheoryGeorge RajnaNo ratings yet
- 7 EnsembleDocument17 pages7 Ensemblevicente aranguizNo ratings yet
- SUMSEM2-2018-19 ECE1006 ETH VL2018199000757 Reference Material I 02-Jun-2019 Statistical Mechanics Bose Einstein and Fermi Dirac DistributionsDocument18 pagesSUMSEM2-2018-19 ECE1006 ETH VL2018199000757 Reference Material I 02-Jun-2019 Statistical Mechanics Bose Einstein and Fermi Dirac Distributionsaghil ashokan aghilNo ratings yet
- Quantum Spin System: 1 GradingDocument4 pagesQuantum Spin System: 1 GradingSouzaSantosNo ratings yet
- Microscopic Study of Magnetostatic Spin WavesDocument3 pagesMicroscopic Study of Magnetostatic Spin WavesErsan HarputluNo ratings yet
- Lec 1Document8 pagesLec 1Atul PandeyNo ratings yet
- Partitionfunction 1Document7 pagesPartitionfunction 1api-230248426No ratings yet
- 5 ThermodynamicsDocument16 pages5 Thermodynamicsabmusic711No ratings yet
- Yuki Endo and Tetsuro Nikuni - Population Dynamics of A Spin-1 Bose Gas Above The Bose-Einstein Transition TemperatureDocument6 pagesYuki Endo and Tetsuro Nikuni - Population Dynamics of A Spin-1 Bose Gas Above The Bose-Einstein Transition TemperaturePomac232No ratings yet
- Partition FunctionDocument7 pagesPartition FunctionAravind Rao KaranamNo ratings yet
- Thermodynamics: Teacher: Shannon SmithDocument71 pagesThermodynamics: Teacher: Shannon SmithShannon SmithNo ratings yet
- SS Lectures MazDocument23 pagesSS Lectures Mazilia1999No ratings yet
- Sect 2Document25 pagesSect 2Amol MahajanNo ratings yet
- 0143-0807 - 22 - 5 - 303jurnal MekstatDocument4 pages0143-0807 - 22 - 5 - 303jurnal MekstatNita HandayaniNo ratings yet
- 22.05 Reactor Physics - Part Three: Useful ToolsDocument5 pages22.05 Reactor Physics - Part Three: Useful ToolsmsakowskNo ratings yet
- Enthalpy, EntropyDocument4 pagesEnthalpy, Entropymanjeet.csedNo ratings yet
- Superconductivity, Superfluidity and Bose-Einstein CondensationDocument21 pagesSuperconductivity, Superfluidity and Bose-Einstein CondensationKomal SahNo ratings yet
- Physics Extra QDocument18 pagesPhysics Extra QGadisNovelNo ratings yet
- PHY4311 Part 1 PDFDocument30 pagesPHY4311 Part 1 PDFHassan Badiru AbdulsalamNo ratings yet
- Theoretical Calculation of The Heat CapacityDocument13 pagesTheoretical Calculation of The Heat Capacityprakush01975225403No ratings yet
- Introduction To Solid State PhysicsDocument80 pagesIntroduction To Solid State Physics林忠佑No ratings yet
- Users Manual: Resistivity of Semiconductors by Four Probe Method at Different TemperaturesDocument23 pagesUsers Manual: Resistivity of Semiconductors by Four Probe Method at Different TemperaturesRobin ChadhaNo ratings yet
- Lecture 10: Quantum Statistical Mechanics: DQDP Which Is Formally Innite. WeDocument11 pagesLecture 10: Quantum Statistical Mechanics: DQDP Which Is Formally Innite. WeSadiq IbrahimNo ratings yet
- Dimensional Analysis Lecture (Cambridge)Document13 pagesDimensional Analysis Lecture (Cambridge)ucaptd3No ratings yet
- 2.57 Nano-to-Macro Transport Processes Fall 2004: V L KK VKK N VDocument7 pages2.57 Nano-to-Macro Transport Processes Fall 2004: V L KK VKK N VcaptainhassNo ratings yet
- Plasma Spectroscopy Inferences From Line Emission: Profiles Not Often Suitable For Inversion, E.G. HollowDocument21 pagesPlasma Spectroscopy Inferences From Line Emission: Profiles Not Often Suitable For Inversion, E.G. HollowkurakidNo ratings yet
- Concepts of Nuclear Medicine Volume I: Concepts of Nuclear Medicine, #1From EverandConcepts of Nuclear Medicine Volume I: Concepts of Nuclear Medicine, #1No ratings yet
- A-Level Chemistry Revision: Cheeky Revision ShortcutsFrom EverandA-Level Chemistry Revision: Cheeky Revision ShortcutsRating: 4 out of 5 stars4/5 (5)
- Mathematical Solution Unifying the Four Fundamental Forces in NatureFrom EverandMathematical Solution Unifying the Four Fundamental Forces in NatureNo ratings yet
- Part 7 Mean Field TheoryDocument40 pagesPart 7 Mean Field TheoryOmegaUserNo ratings yet
- Tensor AnalysisDocument29 pagesTensor AnalysisPugazhenthi Thananjayan100% (2)
- Math 404 Lecture NotesDocument31 pagesMath 404 Lecture NotesOmegaUserNo ratings yet
- Part 2 Basics of Statistical Physics1Document24 pagesPart 2 Basics of Statistical Physics1OmegaUserNo ratings yet
- Part 5 Momentum HopsDocument27 pagesPart 5 Momentum HopsOmegaUserNo ratings yet
- Fundamentals of Statistical Physics: Leo P. Kadanoff University of Chicago, USADocument17 pagesFundamentals of Statistical Physics: Leo P. Kadanoff University of Chicago, USAOmegaUserNo ratings yet
- Outline For Fundamentals of Statistical PhysicsDocument13 pagesOutline For Fundamentals of Statistical PhysicsOmegaUserNo ratings yet
- Part 4 Diffusion and HopsDocument24 pagesPart 4 Diffusion and HopsOmegaUserNo ratings yet
- Part 3 Lattice Quantum Ising RGDocument28 pagesPart 3 Lattice Quantum Ising RGOmegaUserNo ratings yet
- Part 6 Boson-FermionDocument16 pagesPart 6 Boson-FermionOmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L28Document5 pagesStatistical Mechanics Lecture Notes (2006), L28OmegaUserNo ratings yet
- Part 8 Beyond Mean FieldDocument42 pagesPart 8 Beyond Mean FieldOmegaUserNo ratings yet
- Chapter 1 ChemDocument4 pagesChapter 1 ChemOmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L14Document4 pagesStatistical Mechanics Lecture Notes (2006), L14OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L20Document4 pagesStatistical Mechanics Lecture Notes (2006), L20OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L30Document4 pagesStatistical Mechanics Lecture Notes (2006), L30OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L13Document7 pagesStatistical Mechanics Lecture Notes (2006), L13OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L21Document4 pagesStatistical Mechanics Lecture Notes (2006), L21OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L19Document3 pagesStatistical Mechanics Lecture Notes (2006), L19OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L15Document4 pagesStatistical Mechanics Lecture Notes (2006), L15OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L7Document8 pagesStatistical Mechanics Lecture Notes (2006), L7OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L16Document4 pagesStatistical Mechanics Lecture Notes (2006), L16OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L26Document5 pagesStatistical Mechanics Lecture Notes (2006), L26OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L5Document11 pagesStatistical Mechanics Lecture Notes (2006), L5OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L3Document5 pagesStatistical Mechanics Lecture Notes (2006), L3OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L18Document6 pagesStatistical Mechanics Lecture Notes (2006), L18OmegaUserNo ratings yet
- Statistical Mechanics Lecture Notes (2006), L12Document7 pagesStatistical Mechanics Lecture Notes (2006), L12OmegaUserNo ratings yet
- Webinar MacDrain ENGDocument45 pagesWebinar MacDrain ENGIrcil Rey QuiblatNo ratings yet
- Umat DocumentationDocument47 pagesUmat DocumentationNgoc-Trung NguyenNo ratings yet
- Exp 4Document6 pagesExp 4RiazNo ratings yet
- Exxonmobil Premium Afme 200 Fact SheetDocument1 pageExxonmobil Premium Afme 200 Fact SheetMahad AbdiNo ratings yet
- Supermix PC550 PDFDocument2 pagesSupermix PC550 PDFmanavNo ratings yet
- Multiple Choice Question AnswersDocument4 pagesMultiple Choice Question AnswersAnkit kannojia100% (1)
- Chemical Analysis of WaterDocument4 pagesChemical Analysis of WatergenciNo ratings yet
- Tutorial 2 NMJ10103 Answer SchemeDocument5 pagesTutorial 2 NMJ10103 Answer SchemeWendy LohNo ratings yet
- SlumpDocument2 pagesSlumpErikson MoralesNo ratings yet
- Kiln Area Learning ReportDocument24 pagesKiln Area Learning ReportAbasiemekaNo ratings yet
- The Role of Microbiology in The Design and Development of Pharmaceutical Manufacturing ProcessesDocument4 pagesThe Role of Microbiology in The Design and Development of Pharmaceutical Manufacturing ProcessesHari RamNo ratings yet
- Pharmaceutical Applications of Zinc Oxide Nanoparticles - A ReviewDocument2 pagesPharmaceutical Applications of Zinc Oxide Nanoparticles - A Reviewhanady2211No ratings yet
- Modern Physics 1Document18 pagesModern Physics 1Harsh GuptaNo ratings yet
- Draft Plant Design PaperDocument65 pagesDraft Plant Design Paper202040336No ratings yet
- Organometallic Chemistry: An Overview of Structures and ReactionsDocument28 pagesOrganometallic Chemistry: An Overview of Structures and ReactionsPadyala SriramNo ratings yet
- An Assignment On Human Physiology.Document7 pagesAn Assignment On Human Physiology.Samira RahmanNo ratings yet
- Ion Selective ElectrodeDocument9 pagesIon Selective ElectrodeRekhaNo ratings yet
- Plant Rarity Lab ReportDocument15 pagesPlant Rarity Lab Reportapi-529961637No ratings yet
- Multi Purpose Floor Coating - Nippon PaintDocument2 pagesMulti Purpose Floor Coating - Nippon PaintNippon Paint Total Coating and Construction SolutionsNo ratings yet
- A Lead AcetateDocument4 pagesA Lead AcetateInarat HussainNo ratings yet
- LEARNING ACTIVITY SHEET-CHEM 1 q1 Week 4Document8 pagesLEARNING ACTIVITY SHEET-CHEM 1 q1 Week 4Jhude JosephNo ratings yet
- Engineering Fracture Mechanics Prof. K. Ramesh Department of Applied Mechanics Indian Institute of Technology, MadrasDocument24 pagesEngineering Fracture Mechanics Prof. K. Ramesh Department of Applied Mechanics Indian Institute of Technology, MadrasAbhishek AroraNo ratings yet
- MET DOC-Installation Rev0Document11 pagesMET DOC-Installation Rev0IonCube KhanzNo ratings yet
- Cleaning Validation in Pharmaceutical IndustriesDocument5 pagesCleaning Validation in Pharmaceutical IndustriesAbhishek RajNo ratings yet
- Mole RatioDocument12 pagesMole Ratiomaysanati2007No ratings yet
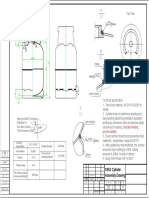
- 10KG Cylinder Assembly DrawingDocument1 page10KG Cylinder Assembly Drawingpaul akhanobaNo ratings yet
- s28170-26 (Lincoln)Document51 pagess28170-26 (Lincoln)hanz bermejoNo ratings yet
- Make The Grade As and A2 Chemistry Chemistry Revision Guide Edexcel As A2 Modular Nelson Advanced ScienceDocument147 pagesMake The Grade As and A2 Chemistry Chemistry Revision Guide Edexcel As A2 Modular Nelson Advanced Scienceareyouthere92100% (3)
- 3196 I FRAME BulletinDocument24 pages3196 I FRAME BulletinGILBERTO YOSHIDANo ratings yet