Professional Documents
Culture Documents
Optical Properties of ZnO Nanostructures - Reviews (RIP)
Optical Properties of ZnO Nanostructures - Reviews (RIP)
Uploaded by
KỲ DUYÊNOriginal Description:
Original Title
Copyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
Optical Properties of ZnO Nanostructures - Reviews (RIP)
Optical Properties of ZnO Nanostructures - Reviews (RIP)
Uploaded by
KỲ DUYÊNCopyright:
Available Formats
Zinc oxide
DOI: 10.1002/smll.200600134
Optical Properties of ZnO Nanostructures
Aleksandra B. Djurisic* and Yu Hang Leung
From the Contents
1. Introduction............. 945
2. Spontaneous Emission
................................ 947
3. Stimulated Emission 951
4. Nonlinear Optical
Properties................ 955
5. Optical Properties of
Doped ZnO............... 956
6. Conclusions and
Outlook.................... 957
Keywords:
nanostructures
photoluminescence
spontaneous emission
stimulated emission
zinc oxide
Many morphological variations of nanostructured ZnO lead to some interesting optical properties.
944 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
We present a review of current research on the optical properties of ZnO
nanostructures. We provide a brief introduction to different fabrication
methods for various ZnO nanostructures and some general guidelines on
how fabrication parameters (temperature, vapor-phase versus solution-phase
deposition, etc.) affect their properties. A detailed discussion of photo-
luminescence, both in the UV region and in the visible spectral range, is
provided. In addition, different gain (excitonic versus electron hole plasma)
and feedback (random lasing versus individual nanostructures functioning as
FabryPerot resonators) mechanisms for achieving stimulated emission are
described. The factors affecting the achievement of stimulated emission are
discussed, and the results of time-resolved studies of stimulated emission are
summarized. Then, results of nonlinear optical studies, such as second-
harmonic generation, are presented. Optical properties of doped ZnO
nanostructures are also discussed, along with a concluding outlook for
research into the optical properties of ZnO.
1. Introduction
Zinc oxide is a material with great potential for a variety
of practical applications, such as piezoelectric transducers,
optical waveguides, surface acoustic wave devices, varistors,
phosphors, transparent conductive oxides, chemical and gas
sensors, spin functional devices, and UV-light emitters.
[1, 2]
Its wide bandgap ( 3.37 eV at room temperature
[1]
) makes
ZnO a promising material for photonic applications in the
UV or blue spectral range, while the high exciton-binding
energy (60 meV)
[1]
allows efficient excitonic emission even
at room temperature. In addition, ZnO doped with transi-
tion metals shows great promise for spintronic applica-
tions.
[3]
It has also been suggested that ZnO exhibits sensi-
tivity to various gas species, namely ethanol (C
2
H
5
OH), ace-
tylene (C
2
H
2
), and carbon monoxide (CO), which makes it
suitable for sensing applications. Moreover, its piezoelectric
property (originating from its non-centrosymmetric struc-
ture) makes it suitable for electromechanical sensor or ac-
tuator applications. Also, ZnO is biocompatible which
makes it suitable for biomedical applications. Last but not
least, ZnO is a chemically stable and environmentally
friendly material. Consequently, there is considerable inter-
est in studying ZnO in the form of powders, single crystals,
thin films, or nanostructures.
A variety of ZnO nanostructure morphologies, such as
nanowires,
[49]
nanorods,
[1014]
tetrapods,
[1418]
and nanorib-
bons/belts,
[6, 1821]
have been reported. ZnO nanostructures
have been fabricated by various methods, such as thermal
evaporation,
[46, 9, 1421]
metalorganic vapor phase epitaxy
(MOVPE),
[12]
laser ablation,
[13]
hydrothermal synthesis,
[7, 10, 11]
and template-based synthesis.
[8]
Recently, novel morpholo-
gies such as hierarchical nanostructures,
[22]
bridge-/nail-like
nanostructures,
[23]
tubular nanostructures,
[24]
nanosheets,
[25]
nanopropeller arrays,
[26, 27]
nanohelixes,
[26, 28]
and nano-
rings
[26, 28]
have, amongst others, been demonstrated. Some
of the possible ZnO nanostructure morphologies are shown
in Figures 13. Several recent review articles have summar-
ized progress in the growth and applications of ZnO nano-
structures.
[2931]
The growth and properties of ZnO nano-
structures have been extensively studied,
[3275]
but there are
still a number of unanswered questions concerning the rela-
tionship between fabrication conditions and optical proper-
ties.
The fabrication methods for ZnO nanostructures can be
divided into two groups: spontaneous growth and template-
based synthesis (for example, using an alumina template).
Fabrication without a template can occur either by using
metal catalysts or may be self-catalyzed. The use of metal
catalysts, such as Au, can be an advantage for achieving
aligned and selective area growth.
[4]
Aligned nanorods can
also be obtained by a hydrothermal method without any
metal catalyst.
[7, 46]
The degree of alignment and the ach-
ieved aspect ratio was dependent on the seed layer used
and the fabrication conditions.
[7, 46]
An improvement in
alignment of the rods perpendicular to the substrate was ob-
tained when zinc acetate was used to prepare the nanocrys-
talline seed layer instead of ZnO nanoparticles.
[7]
The
growth of ZnO by vapor deposition is typically affected by
temperatures of the source and the substrate, the distance
between the source and the substrate, the heating rate, the
gas flow rate, tube diameter, and the starting precursor(s).
[75]
The influence of these factors on ZnO morphology was
[*] Dr. A. B. Djurisic
Department of Physics, The University of Hong Kong
Pokfulam Road (Hong Kong)
Fax: (+852) 2559-9152
E-mail : dalek@hkusua.hku.hk
Y. H. Leung
Department of Chemistry, The University of Hong Kong
Pokfulam Road (Hong Kong)
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim 945
Optical Properties of ZnO Nanostructures
studied in detail recently,
[75]
but the effects of these factors
on the optical properties of fabricated nanostructures are
still unknown. Different experimental conditions, such as
for example, solution concentration, temperature, and sub-
strate pretreatment, also affect the growth of ZnO by hy-
drothermal methods. Due to the low growth temperature
(typically under 1008C), the crystalline quality of such sam-
ples is often lower than those fabricated by vapor deposi-
Figure 1. af) Representative scanning electron microscopy images of
various ZnO nanostructure morphologies.
Figure 2. ad) Representative scanning electron microscopy images
of ZnO nanopropeller arrays. Reprinted with permission from
Ref. [27].
Figure 3. AD) Representative scanning electron microscopy images
of ZnO helical nanobelts. Reprinted with permission from Ref. [28].
Aleksandra B. Djurisic obtained her PhD
degree in electrical engineering from the
School of Electrical Engineering at the
University of Belgrade (now Serbia) in
1997. After finishing her PhD studies,
she worked as a postdoctoral fellow at
University of Hong Kong and as an
Alexander von Humboldt postdoctoral
fellow at TU Dresden. She has been as-
sistant professor in the Dept. of Physics
at the University of Hong Kong since
2003. Her research interests include the
optical properties of materials, nano-
materials, wide-bandgap semiconduc-
tors, block copolymers, and opto-
electronic devices.
Y. H. Leung obtained his B.Eng. degree
from the City University of Hong Kong in
2003 and his M. Phil. degree from the
University of Hong Kong in 2005. He is
currently working as a research assistant
at the University of Hong Kong. His re-
search interests include the fabrication,
characterization, and applications of ZnO
nanostructures.
946 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
tion. However, the optical quality of the samples can be im-
proved by annealing under appropriate conditions.
[46]
In this Review, we provide a detailed overview on the
optical properties of ZnO nanostructures. The Review is or-
ganized as follows: In the next section, we discuss spontane-
ous emission from ZnO nanostructures. Low-temperature
and room-temperature photoluminescence in the UV and
visible spectral regions are discussed. Next, an overview of
stimulated emission in various ZnO nanostructures is pro-
vided. In chapter 4, the nonlinear optical properties of ZnO
are presented. Finally, some conclusions and an outlook for
the future are given.
2. Spontaneous Emission
Optical properties of a variety of forms of ZnO, includ-
ing ZnO nanostructures, have been studied by photolumi-
nescence (PL) spectroscopy.
[32122]
The majority of the re-
ported luminescence spectra of ZnO nanostructures have
been measured at room temperature, although variable-tem-
perature photoluminescence studies have been performed
on some of the samples.
[3246]
Room-temperature PL spectra
of ZnO typically consist of a UV emission and possibly one
or more visible bands due to defects and/or impurities.
2.1. UV Emission
Low-temperature photoluminescence measurements of
different nanostructures, such as nanowire/nanowall sys-
tems,
[32]
nanosheets,
[33]
nanowalls,
[44]
nanowires,
[34, 43, 45]
nano-
rods,
[35, 37, 39, 46]
faceted nanorods,
[41]
nanoparticles,
[42]
and
nanoblades and nanoflowers,
[40]
have been reported. Low-
temperature (410 K) PL spectra of ZnO typically exhibit
several peaks (labeled I
0
I
11
), which correspond to bound
excitons.
[76]
An example of a low-temperature PL spectrum
of a ZnO sample exhibiting a number of bound-exciton
peaks is shown in Figure 4. The number of observed bound-
exciton peaks in ZnO nanostructures is typically lower than
that in ZnO single crystals. An example of a low-tempera-
ture PL spectrum of highly faceted ZnO rods with good
crystalline quality
[41]
is shown in Figure 5. Since the relative
intensity of the bound-exciton peaks varies from sample to
sample due to variations in donor/acceptor concentrations
and their capture cross sections,
[77]
variable-temperature PL
measurements can provide useful information about the op-
tical and structural properties of ZnO. However, the assign-
ment of the bound-exciton peaks in ZnO is, in general, con-
troversial for all forms of the samples, namely, ZnO single
crystals, epitaxial films, and nanostructures. For example, it
was proposed that the emission lines I
5
to I
11
in the lower
part of the energy spectrum can be attributed to excitons
bound to neutral acceptors.
[78]
However, other reports in the
literature attributed some of these lines to donor bound ex-
citons.
[76, 79]
The chemical identity of the donors and accept-
ors responsible for different bound-exciton lines still re-
mains unclear (for a complete list of the bound-exciton
peaks generally observed in ZnO, and a summary of the
possible identification of the donors and acceptors, see
Refs. [1, 76]).
One of the commonly observed bound-exciton lines in
ZnO nanostructures is the I
4
line at 3.3628 eV.
[41, 82]
This
emission is typically attributed to the donor bound exciton,
and the donor has been identified as hydrogen.
[76, 80, 81]
Theo-
retical calculations predict hydrogen to be a shallow donor
in ZnO
[123]
and it is reasonable to expect that an uninten-
tional incorporation of hydrogen could frequently happen in
ZnO nanostructure synthesis. While in general there is a
consensus in assigning the I
4
line to hydrogen donors,
[76, 80, 81]
the chemical identity of donors responsible for other donor
bound-exciton lines remains unclear.
For the acceptor bound excitons, the most commonly re-
ported peak is located at 3.3564 eV.
[77]
This peak is common-
ly attributed to excitons bound to Na or Li acceptors.
[1]
Alkali metals are predicted to produce shallow acceptors on
the cation site, but the experimental results demonstrate
that doping with group 1 ions produces complex results.
[124]
Figure 4. Bound-excitonic region of the 10 K PL spectrum for the
forming gas annealed ZnO substrate. Reprinted with permission from
Ref. [77].
Figure 5. PL spectra from ZnO faceted rods at different temperatures
as a function of a) wavelength, and b) energy. The inset shows an
enlarged region of the PL spectrum at 7 K. Reproduced from
Ref. [41].
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 947
Optical Properties of ZnO Nanostructures
However, other acceptor levels have also been proposed,
such as an acceptor complex involving a N impurity on an
O site.
[81]
However, some authors attribute this line to a
donor bound exciton instead.
[76, 79]
Bound-exciton lines I
6
, I
8
,
and I
9
have been assigned to excitons bound to Al, Ga, and
In donors, respectively.
[76]
On the other hand, Thonke
et al.
[82]
proposed that the weak 3.357 eV line corresponds
to the acceptor bound exciton, while the I
8
line at 3.3597 eV
was found to be a donor bound-exciton line.
In addition to commonly observed acceptor bound-exci-
ton lines, emission at 3.332 eV (labeled as I
a
) was recently
observed in low-temperature PL spectra of ZnO epilayers
grown on CaF
2
(111).
[109]
Since this peak occurs in the spec-
tral region where two-electron satellites (TES) of donor
bound-exciton peaks are expected to occur,
[82]
careful ex-
amination of the peak position in respect to known bound-
exciton positions and expected TES peaks (see Ref. [76] for
the positions of TES lines for different bound-exciton
peaks) is necessary. In addition, the occurrence of peak near
3.333 eV may indicate excitons bound to structural de-
fects.
[76]
Therefore, further work is needed for conclusive
identification of the origin of different bound-exciton lines
in ZnO. The assignment of several bound-exciton lines, es-
pecially I
9
, is still controversial and conclusive chemical
identification of the majority of donors and acceptors has
not been accomplished.
At low temperatures, in addition to bound-exciton
peaks, two-electron satellite transitions can be observed in
the spectral region 3.323.34 eV.
[77]
These transitions corre-
spond to a radiative recombination of donor bound excitons,
which leaves the donor in an excited state. Thus, they are lo-
cated at an energy lower by an amount equal to the differ-
ence between the first excited and ground states of the
donor, so that their position in respect to the donor bound-
exciton peaks can be used to estimate donor-binding ener-
gies.
[82, 83]
Finally, low-temperature PL spectra can also contain
donoracceptor pair transitions and longitudinal optical
(LO) phonon replicas.
[77]
First-, second-, and third-order LO
phonon replicas can typically be observed.
[44]
The LO
phonon energy can be determined from the separation be-
tween the exciton peaks and their LO phonon replicas, and
for ZnO it is 7173 meV.
[45, 84]
Since donoracceptor pair
transitions and some of the LO phonon replicas occur in the
same spectral region (3.2183.223 eV),
[77]
care needs to be
taken in assigning the peaks observed in this region. In addi-
tion, two-phonon replicas due to two transverse optical pho-
nons (separation of 108 meV) were also reported in ZnO
thin films prepared by spray pyrolysis.
[113]
With regard to the temperature dependence of the ob-
served peaks, a red shift of the free-exciton emission with
increasing temperature occurs. The intensity of the bound-
exciton peaks and the LO phonon replicas decreases with
increasing temperature, and only free-exciton emission can
be observed at room temperature. In ZnO epilayers, free-
exciton emission was found to dominate the spectra above
70 K.
[109]
Similar behavior, with the disappearance of
bound-exciton peaks above 150 K, was also observed in
ZnO single-crystal samples.
[110]
The bound-exciton line for
ZnO nanoparticles embedded in alkali halide crystals also
disappeared at 125 K.
[115]
The exact temperature at which
the bound-exciton line will disappear depends on the identi-
ty of the donors or acceptors, since different donors/accept-
ors will be thermally ionized at different temperatures. It
should be noted that in the case of donoracceptor pair
transition, disappearance of this peak with increasing tem-
perature can be accompanied by the appearance of acceptor
bound-exciton peaks if the acceptors are thermally ionized
at higher temperature than the donors.
[117]
However, what all ZnO samples (single crystals, films,
and nanostructures) have in common is the disappearance
of bound-exciton peaks at temperatures in the range 50
150 K, while at room temperature only free-exciton emis-
sion is observed. The presence of free-exciton emission at
low temperatures, as well as a distinction between A and B
exciton peaks, is usually considered to indicate high quality
in ZnO samples.
[111]
It should be noted that this criterion for
sample quality is less arbitrary than the ratio between UV
and defect emission, which is sometimes used to estimate
sample quality,
[116, 121]
and which is dependent on excitation
area and power.
[74]
Distinction between A and B exciton
peaks is usually not possible above 160 K,
[77]
and the
phonon replicas typically cannot be clearly resolved above
250 K.
[44]
In very-high-quality samples, higher-order exci-
ton lines can also be observed at low temperatures.
[118, 122]
Biexciton emission was observed at 77 K in high-quality
epitaxial ZnO films.
[111]
The biexciton binding energy was
estimated to be 15 meV.
[111]
Biexciton emission has also
been observed in ZnO nanowires
[112]
and nanorods.
[35, 114]
The obtained energy separation between exciton and biexci-
ton peaks in ZnO nanowires was 20 meV, in good agree-
ment with the results obtained on other forms of ZnO.
[112]
The reported energy separation in the case of nanorods was
18 meV,
[35]
and biexciton emission persisted up to
200 K.
[114]
Clear observation of free-exciton and biexciton
lines at low temperatures is usually considered as an indica-
tion of very good sample quality.
[35]
In room-temperature PL spectra, some variation of the
position of the PL peak can be observed for different nano-
structures. This is illustrated in Figure 6, where different UV
Figure 6. Room-temperature PL spectra of various nanostructures in
the UV range: 1) Tetrapods, 2) needles, 3) nanorods, 4) shells,
5) highly faceted rods, 6) ribbons/combs.
948 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
peak positions (387 nm for tetrapods, 381 nm for needles,
397 nm for nanorods, 377 nm for shells, 379 nm for faceted
rods, and 385.5 nm for ribbons/combs) can be observed.
Room-temperature band-edge emission in ZnO nanostruc-
tures was reported to occur at 373,
[49]
378,
[46, 53, 57]
380,
[47, 64, 67]
381,
[55]
383,
[52, 62]
384391,
[56]
387.5,
[58]
389,
[50, 62]
and 390 nm.
[59]
Individual nanostructures, such as nanobelts, exhibited UV
emission in a range between 384 and 391 nm.
[56]
These dif-
ferences in the peak positions of individual nanobelts, which
are sufficiently large so that there could be no quantum con-
finement effects, indicate that there is likely a different ex-
planation for the variation in the band-edge emission in
ZnO nanostructures reported in different studies. Even
though quantum confinement has been proposed as a cause
of the blue shift of the band-edge emission with decreasing
size,
[69]
any shift due to quantum confinement in nanocrys-
tals with diameters of 57, 38, and 24 nm is not likely consid-
ering the fact that the Bohr radius of ZnO is 2.34 nm.
[70]
One possible reason for the variations in the position of
the band-edge emission in various ZnO nanostructures with
relatively large dimensions are different concentrations of
native defects. Since the defect density on the surface is
higher than in the bulk,
[125]
spectral shifts due to different
defect concentrations are expected to occur in nanostruc-
tures with different sizes due to different surface-to-volume
ratios. The fact that the decay times in time-resolved PL
from ZnO nanorods are size dependent
[71]
is in agreement
with the assumption of different defect levels/concentrations
for structures with different surface-to-volume ratios. Thus,
the defects could affect the position of the band-edge emis-
sion as well as the shape of the luminescence spectrum. Al-
though there have been several reports with strong UV and
weak defect emission in ZnO nanostructures,
[49, 50]
in some
cases only defect emission is observed
[48]
or the UV emission
is much weaker compared to the defect emission.
[46]
There-
fore, clarifying the origins of different defect emissions is an
important issue. However, it should be noted that the ratio
of the intensity of UV and defect emission is dependent on
the excitation density,
[36, 74]
as well as the excitation area.
[74]
Thus, the ratios of these two emissions cannot be used as an
absolute determining factor of the crystalline quality of
ZnO, although they are useful in comparing the quality of
different samples when the measurements are performed
under identical excitation conditions.
2.2. Defect Emissions
Room-temperature PL spectra from ZnO can exhibit a
number of different peaks in the visible spectral region,
which have been attributed to the defect emission. Emission
lines at 405, 420, 446, 466, 485, 510, 544, 583, and 640 nm
have been reported (see Ref. [36] and references therein).
Several calculations of the native defect levels in ZnO have
been reported,
[8587, 124]
as summarized in Figure 7. An exam-
ple of defect emissions (normalized PL spectra) from differ-
ent ZnO nanostructures is shown in Figure 8.
Green emission is the most commonly observed defect
emission in ZnO nanostructures,
[4, 4749, 52, 53, 5658, 61, 62, 64, 67, 68]
similar to other forms of ZnO. The intensity of the blue
green defect emission was found to be dependent on the
nanowire diameter,
[5, 64]
but both increased
[5]
and de-
creased
[64]
defect emission intensity with decreased wire di-
ameter were reported. Several different hypotheses have
been proposed: Green emission is often attributed to singly
ionized oxygen vacancies,
[4749, 64, 68]
although this assignment
is highly controversial. Other hypotheses include antisite
oxygen,
[56]
which was proposed by Lin et al.
[85]
based on the
band structure calculations. Green emission was also attrib-
uted to oxygen vacancies and zinc interstitials.
[67]
Cu impuri-
ties have been proposed as origin of the green emission in
ZnO.
[88]
Blue-green defect emission was also reported in Cu
doped ZnO nanowires.
[65]
However, although Cu was identi-
fied as a possible cause of green emission in ZnO,
[88]
this
cannot explain the defect emission in all ZnO nanostructure
samples, especially those where defect emission exhibits
strong dependence on annealing temperature and atmos-
phere which would be more consistent with an intrinsic
defect rather than Cu impurity. Other hypotheses include
Figure 8. Room-temperature PL spectra of different nanostructures:
1) Tetrapods, 2) needles, 3) nanorods, 4) shells, 5) highly faceted
rods, 6) ribbons/combs.
Figure 7. Illustration of the calculated defect energy levels in ZnO
from different literature sources (data marked with the subscript a
originate from Ref. [85], those marked with b stem from Ref. [87],
and those marked with c originate from Ref. [86]). V
Zn
, V
Zn
, and
V
Zn
2
denote neutral, singly charged, and doubly charged zinc vacan-
cies, respectively. Zn
i
o
and Zn
i
indicate neutral zinc interstitials,
while Zn
i
+
denotes a singly charged zinc interstial. V
O
o
and V
O
denote
neutral oxygen vacancies, while V
O
+
denotes a singly charged
oxygen vacancy. O
i
represents an oxygen interstitial. V
O
Zn
i
denotes a
complex of an oxygen vacancy and zinc interstitial.
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 949
Optical Properties of ZnO Nanostructures
various transitions related to intrinsic defects, such as
donoracceptor transitions,
[89]
recombination at V
o
** centers
(where these centers are generated by surface trapping of
photogenerated holes, followed by recombination with elec-
tron in an oxygen vacancy V
o
*),
[90, 91]
zinc vacancy,
[92, 93]
and
surface defects.
[36]
Although the singly ionized oxygen va-
cancy
[94]
is a commonly cited hypothesis, which is supported
by reports of the enhancement of the green defect by an-
nealing at temperatures above 6008C (attributed to out-dif-
fusion of O),
[47]
this assignment has been questioned recent-
ly.
[36, 88]
The donoracceptor transition hypothesis used to ex-
plain the green and yellow emissions has also been chal-
lenged.
[95]
On the other hand, while the Zn vacancy hypoth-
esis is supported by the study of the effect of O and Zn
implantation,
[93]
a blue rather than green emission would be
expected based purely on the theoretically predicted energy
levels for Zn vacancy.
[86]
Therefore, the origin of the green
emission is still an open and controversial question and the
identification of the exact origin of this emission requires
further study.
While the type of defect responsible for the green emis-
sion has not yet been conclusively identified, there is con-
vincing evidence that it is located at the surface. It was
shown that coating ZnO nanostructures with a surfactant
suppressed green emission.
[36]
Polarized luminescence ex-
periments from aligned ZnO nanorods also indicated that
green emission originated from the surface of the nano-
rods.
[72]
The surface recombination layer responsible for visi-
ble emission in ZnO nanowires was estimated to be 30 nm
in thickness.
[73]
Also, the possible presence of Zn(OH)
2
at
the surface, especially for nanostructures prepared by solu-
tion methods, could affect the emission spectra from ZnO
nanostructures.
[51]
Yellow defect emission is also commonly reported in
ZnO nanostructures,
[46, 62, 96]
and it represents a common fea-
ture in samples prepared from aqueous solutions of zinc ni-
trate hydrate and hexamethylenetetramine.
[46, 96]
This emis-
sion is typically attributed to an oxygen interstitial,
[46, 92, 96]
al-
though a Li impurity represents another possible candi-
date.
[96]
The deep levels responsible for green and yellow
emissions were found to be different;
[92, 96]
unlike the defect
responsible for the green emission, the defect responsible
for the yellow emission is not located at the surface.
[96]
In addition to green and yellow emissions, orange-red
emissions are often also observed.
[53, 57, 67, 97, 98]
Fan et al.
[53, 57]
reported that the visible emission in ZnO dendritic wires
and nanosheets consisted of two components centered at
540 and 610 nm. The intense visible emission in ZnO
nanosheets was tentatively attributed to surface disloca-
tions.
[57]
Orange-red emission at 626 nm in ZnO nanorods
was attributed to oxygen interstitials.
[67]
In addition, orange
emission at 640650 nm in ZnO needles
[98]
and nano-
wires
[97]
was proposed to be due to oxygen-rich samples, in
agreement with a previous study on ZnO films.
[99]
This emis-
sion could be reduced by annealing under vacuum or in a
H
2
/Ar mixture.
[97]
However, although these treatments
quenched the visible defect emission, near-infrared (NIR)
emission at 750 nm was enhanced.
[97]
It was shown that
green, yellow, and red-NIR emissions originate from differ-
ent types of defects by depth-resolved cathodoluminescence
and PL measurements.
[100]
The NIR and the yellow emis-
sions were found to have different decay properties, and it
was proposed that they involved a similar final state related
to excess oxygen but with different initial states (conduction
band and donor centers).
[101]
It should be noted that al-
though the majority of studies attribute red-NIR emission
to excess oxygen, zinc interstitials were also proposed to ex-
plain the origin of a red emission in ZnO particles.
[102]
Thus,
although this emission is less controversial than the green
one, further studies are needed to clarify its origin.
A similar conclusion applies to other defect emissions
reported in ZnO, such as blue and violet defect emissions.
Zhao et al.
[55]
reported emission at 3.0 eV (413 nm), which
was attributed to a zinc vacancy, while a violet emission in
ZnO nanobelts at 421 nm was attributed to interstitial
zinc.
[56]
Blue emission at 440 nm was reported for tetrapo-
dal nanocrystals,
[59]
while other reports indicate the presence
of both violet (419 nm) and blue (438 nm) emissions in ZnO
tetrapods.
[60]
The violet emission was attributed to interface
traps, while blue emission was attributed to oxygen vacan-
cies.
[60]
A blue emission band ( 420 and 444 nm) in ZnO
nanowires
[63]
and ZnO nanocrystals (at 442 nm)
[68]
was
also attributed to oxygen vacancies. It is obvious that for
many types of defects, different studies assign them different
emission-peak positions. Thus, while the origins of some of
the emission peaks (such as yellow for example) are less
doubtful, the origin of defect emissions in ZnO is still an un-
resolved question in spite of a large number of reports.
In addition to identifying the origin of the defect emis-
sions, an important question is the suppression of defect
emission either by varying the fabrication conditions or by
post-fabrication treatment. It was reported that the green
emission from ZnO nanoparticles can be suppressed by em-
bedding the nanoparticles into a synthetic opal whose pho-
tonic bandgap overlaps with the deep-level emission.
[61]
An-
other way to suppress green defect emission is by coating of
the surface with surfactant.
[36]
Hydrogen plasma was also
shown to enhance UV-to-defect emission intensity ratio for
ZnO nanorods.
[103]
As for the yellow emission, it has been
shown that it can be reduced by annealing in a reducing en-
vironment (hydrogen/argon mixture).
[46]
2.3. Defect Identification using Other Techniques
There are several techniques that can be used combined
with photoluminescence to identify the origin of the defect
emissions in ZnO. A useful technique for identifying para-
magnetic defects is electron paramagnetic resonance (EPR)
spectroscopy, which has been used to study defects in
ZnO.
[36, 87, 93, 95, 104, 126]
For an overview of previous studies see
Ref. [126], while some of the more recent results are given
in Ref. [36]. Similar to the origin of the green luminescence,
the assignment of the peaks in EPR spectra of ZnO has
been controversial. The singly ionized oxygen vacancy hy-
pothesis was proposed by Vanheusden et al.
[92]
based on the
correlation between the EPR peak at g1.96 and green
emission intensity. However, other studies attribute the
950 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
peak at g1.99 to oxygen vacancies.
[88]
Although the assign-
ment of the peaks is in question, this technique is still a
useful tool to obtain more information about the defects,
and its application to nanostructures is straightforward since
nanostructures can be treated as any other sample in pow-
dered form.
Positron annihilation spectroscopy (PAS) has been used
in the past together with photoluminescence to study defect
emissions in ZnO.
[105, 106]
Although no conclusive identifica-
tion of the defects responsible for the visible emissions has
been made, this technique still enables more information to
be obtainined about the defect levels, which helps to at least
eliminate some of the possible candidates for the causes of
defect emission. It should be noted however that although
PAS has been applied to some nanostructured samples, the
interpretation of the data from nanorods or nanowires
would be very complicated due to the voids in the samples.
Another useful technique for studying defect levels is deep-
level transient spectroscopy (DLTS). A DLTS study of ZnO
single crystals established that defects at 0.10, 0.12, 0.29, and
0.59 eV below the conduction band were found,
[107]
although
the defect identity was not conclusively established. Again,
this technique is also more applicable to thin-film or single-
crystal samples. However, it is expected that conclusions
from studying defect emission from ZnO in these forms
could be extrapolated to identify defects in nanostructures
since defect positions and behavior are similar for nano-
structured and bulk samples.
3. Stimulated Emission
Due to its high exciton-binding energy, ZnO is of inter-
est for the achievement of excitonic stimulated emission at
room temperature, which has a lower threshold than elec-
tronhole plasma recombination. While there have been nu-
merous reports on optically pumped lasing and amplified
spontaneous emission from ZnO,
[4, 41, 98, 127168]
no electrically
pumped lasing has been achieved as yet. Amplified sponta-
neous emission was reported for a self-organized network of
ZnO fibers,
[161]
while lasing has been reported in a number
of different structures such as, for example, nanowires,
[5, 138]
tetrapods,
[151154]
and nanoribbons/combs.
[154]
A very broad
range of lasing thresholds has been reported for different
ZnO nanostructures, ranging from 8 kWcm
2
(ZnO
fibers)
[130]
to 867 kWcm
2
(ZnO nanorods).
[163]
In the follow-
ing section, the experimental results of the stimulated emis-
sion in ZnO nanostructures will be summarized. The discus-
sion of the basic principles will include some comparisons
with stimulated emission from other forms of ZnO.
3.1. Gain Mechanism
Stimulated emission in ZnO can be achieved either by
excitonexciton (EE) scattering or electronhole plasma
(EHP) recombination. As the excitation power increases,
sharp peaks will appear in the emission spectra from ZnO
(highly-faceted rods), as illustrated in Figure 9. Due to the
significantly shorter decay time of the stimulated emission
compared to spontaneous emission, lasing peaks can be ob-
served more clearly in time-resolved spectra, as shown in
Figure 10. As the excitation power increases, the increase in
intensity and the appearance of narrow lasing modes can be
observed. With a further increase of excitation power, lasing
in the EHP mode occurs. The lasing in these different exci-
tation regimes will be discussed below.
3.1.1. ExcitonExciton Scattering
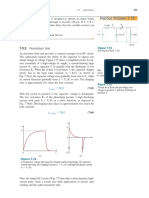
The peak position of the emission resulting from inelas-
tic collisions between excitons is given by:
[167]
E
n
E
ex
E
b
ex
1 1
n
2
3kT=2 1
where n=2, 3,, k is the Boltzmann constant, T is the tem-
perature, and E
b
ex
=60 meV is the exciton binding energy.
Figure 9. Emission spectra from highly faceted ZnO rods at different
excitation powers. The excitation wavelength was 267 nm, and the
pulse duration was 1 ps.
Figure 10. Time-resolved PL spectra for spontaneous emission
(shown at 4 ps), stimulated emission due to excitonexciton scatter-
ing (shown at 8 ps due to a longer delay time), and stimulated emis-
sion due to EHP (shown at 4 ps). Due to the very high intensity emis-
sion in the EHP regime, the other two spectra have been multiplied
by a factor of 10 to improve the clarity of presentation. Reprinted
from Ref. [41].
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 951
Optical Properties of ZnO Nanostructures
The transition from spontaneous emission to stimulated
emission is evidenced by narrowing of the emission with a
full-width half maximum (FWHM) about two orders of
magnitude lower compared to the FWHM of spontaneous
emission.
[156]
The threshold for lasing due to excitonexciton
scattering in nanostructures is typically 23 times
lower
[98, 138, 154]
than that for lasing in the EHP regime.
3.1.2. ElectronHole Plasma
With increasing excitation energy, the density of the ex-
citons in ZnO will also increase. As the exciton density in-
creases, binding energy decreases. The EHP plasma forms
at densities higher than the Mott density, given by:
[1]
n
M
kT
2a
3
B
E
b
ex
2
where a
B
is the Bohr radius. This is estimated to be 3.7
10
19
cm
3
,
[1]
although lower estimates such as 410
18
cm
3
have also been reported.
[138]
The EHP emission is typically
more broad and red shifted compared to emission due to
excitonexciton scattering.
[1]
The red shift of the EHP emis-
sion is the result of bandgap renormalization.
[140]
Coexis-
tence of EE and EHP emissions observed in ZnO thin films
was attributed to spatial nonuniformity of the sample and
the beam profile.
[127]
However, time-resolved studies on dif-
ferent ZnO nanostructures indicate that the coexistence
may originate from the fast decay of EHP emission since a
blue shift of the emission with time and eventual bandgap
recovery could be clearly observed in time-resolved spec-
tra.
[154]
The evolution of the lasing spectra with an increase
of excitation power has been studied in detail for single
ZnO nanowires.
[138]
3.2. Feedback Mechanism
The coherent feedback in ZnO nanostructures or nano-
structured films can be provided by two basic mechanisms,
as illustrated in Figure 11. In the first case, coherent feed-
back is provided by multiple reflections from the end facets
of the nanostructure, which serves as a FabryPerot resona-
tor. It has been shown that UV emission is typically en-
hanced at the ends of the nanowire, while defect (green)
emission is emitted from all parts of the nanowire,
[138]
as
shown in Figure 12. In the second case, the coherent feed-
back is provided by multiple scattering events. An example
of random lasing from ZnO is illustrated in Figure 13.
Random lasing and individual nanostructures as Fabry
Perot resonators are discussed in detail below.
3.2.1. Random Lasing
In random lasers, coherent feedback is provided by re-
curring scattering events.
[169]
Cavities are self-formed, and
the main requirement to observe this type of lasing is that
the scatterer size is smaller than the emission wave-
length.
[162]
The dependence of the lasing on the excitation
area has been demonstrated in ZnO polycrystalline films,
[128]
nanorods,
[133]
and nanowires,
[145]
and the closed loops along
which lasing occurred have been observed.
[128]
Random
Figure 11. Illustration of two different feedback mechanisms:
a) Nanostructures as FabryPerot resonators, b) a random laser.
Figure 12. Images of green/UV PL: a) Topographic, b) UV, and
c) green near-field PL images. Scale bar =5 mm. d) Far-field image of
green/UV PL showing enhancement of the UV PL near the end of the
wire. Scale bar =5 mm. Reprinted with permission from Ref. [138].
952 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
lasing in ZnO was reported in polycrystalline thin
films,
[128, 143, 157, 159]
nanoparticles,
[155, 160]
ZnO powder films,
[162]
nanorods,
[133, 143]
nanoneedles,
[156]
and nanowires.
[145]
The film
structure and crystallinity were found to affect the lasing
threshold,
[143]
and the lasing characteristics were also found
to be dependent on the strain.
[157]
Random lasing was also
demonstrated from thin-film ZnO ridge waveguides, with
[159]
and without
[159]
a MgO capping layer. The lasing threshold
in ZnO nanoneedle random lasers was found to be signifi-
cantly lower than the reported value for conventional lasing
in ZnO nanocolumns.
[155]
One distinguishing characteristic of a random laser is
that stimulated emission can be observed in all directions,
and the mode structure in the measured spectra shows angu-
lar dependence.
[162]
While the interpretation of the data is
straightforward in the case of polycrystalline films, for the
case of a nanowire ensemble with random nanowire orienta-
tion, it is difficult to establish from angular-dependence
measurements of the emission spectra whether the feedback
originates from individual wires acting as resonators or from
scattering between the wires. Another distinguishing charac-
teristic of a random laser is that the lasing threshold de-
pends on the excitation area.
[162]
The lasing threshold in-
creases as the excitation area decreases, and eventually no
lasing can be observed in areas smaller than a critical
size.
[162]
However, careful analysis of the obtained data is
needed when measurements are performed on an ensemble
of nanostructures instead of polycrystalline films. Since dif-
ferent nanostructures can
have different lasing thresh-
olds,
[138]
reduction in a lasing
threshold observed with in-
creased excitation area could
simply be a result of an indi-
vidual nanostructure with a
lower threshold becoming
excited.
One possible way to dis-
tinguish between the two
possible feedback mecha-
nisms is to analyze the Fouri-
er transform of the lasing
spectrum.
[145]
In order to dis-
tinguish between the possi-
bility of having multiple
closed loops and multiple
lasing modes in individual
FabryPerot cavities formed
by crystal facets of the nano-
structrures, it may be neces-
sary to observe where the
lasing originates from, as de-
scribed elsewhere.
[128]
Careful
interpretation of the lasing
data is necessary to conclu-
sively establish the feedback
mechanism. Although
random lasing has been pro-
posed to explain stimulated
emission from short needles with sharp top surfaces,
[156]
stimulated emission was observed from individual, relatively
short nanorods with pyramidal tops indicating that multiple
scattering was not necessary to achieve stimulated emis-
sion.
[166]
3.2.2. Nanostructures as FabryPerot Resonators
Lasing from ZnO nanowires with each wire forming a
FabryPerot resonator bound by reflecting (0001) facets was
first reported by Huang et al.
[4]
Since then, there have been
numerous reports on stimulated emission from various ZnO
nanostructures. Lasing was reported in microtubes,
[129, 150]
nanocoral reefs and nanofibers,
[130]
whiskers,
[131]
nano-
wires,
[4, 138, 139, 147149]
nanorods,
[132, 134, 154]
nanoribbons,
[135, 136, 139]
nanocombs,
[137, 154]
and tetrapod nanostructures.
[151154, 168]
While some of the measurements have been performed on
nanostructure ensembles, stimulated emission from individ-
ual nanostructures
[135, 136, 138, 139, 151, 152]
as well as nanostructures
dispersed with very low density (<10 per laser spot)
[168]
was
also obtained, clearly demonstrating that in those cases the
feedback could not be obtained from multiple random scat-
tering. While in some cases, such as individual nanowires
[138]
and nanoribbons,
[138, 139]
the identification of the cavity is
straightforward, lasing has also been demonstrated with
nanostructures with more complex morphologies. Also,
stimulated emission was obtained not only for nanostruc-
tures fabricated at high temperatures by vapor deposi-
Figure 13. a) Schematic diagram of the laser measurement setup. b) Light curves for the samples after
various ion-irradiation times. The inset shows the maximum emission intensity of the TE mode as a func-
tion of polarization angle. c) Emission spectrum of the as-grown ZnO thin film under a pump power of
1.6 MWcm
2
. d) Evolution of emission spectra of the irradiated sample (30 min) under different pump
intensities. e) Emission spectrum of the sample irradiated for 60 min under a pump power of
1.6 MWcm
2
. Reprinted with permission from Ref. [156] .
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 953
Optical Properties of ZnO Nanostructures
tion,
[131, 137, 154, 168]
but also for nano-
structures fabricated at low tem-
perature from aqueous solu-
tion.
[134, 154]
Detailed mode analysis of
nanostructures as laser cavities has
been performed.
[138, 153]
Good
agreement in ZnO nanowires be-
tween the measured modes with
similar polarization and expected
mode spacing is given by:
[138]
Dl
l
2
2L n l
dn
dl
3
where Dl is the mode spacing, l is
the wavelength, n is the refractive
index, and L is the length. The
linewidth of the measured lasing
modes was also in good agreement
from the theoretical estimate of
the order of 1 nm obtained for a
FabryPerot resonator by using the
following expression:
[138]
Dn
c
4pLn
ln R
1
R
2
1 T
i
2
4
where c is the speed of light, R
1
and R
2
are the reflectivities of the
mirrors, and T
i
is the transmittance
of the internal medium of the
cavity. The threshold gain can be
expressed as:
[135]
g
th
a
1
2L
ln
1
R
1
R
2
5
where L is the length and a is the absorption loss. It should
be noted, however, that thresholds in individual nanowires
can vary by orders of magnitude, which was attributed to
differences in dimensions, the condition of the cavity, and
the extent of substrate coupling.
[138]
The threshold may also
be affected by damage of the nanowire during transfer from
the growth substrate to the support substrate for the meas-
urements.
[153]
Among other more complicated cavities, ZnO tetrapods
have been most thoroughly studied.
[151154, 168]
The lasing
threshold and mode structure of a ZnO tetrapod were
found to be dependent on the tetrapod position on the sub-
strate (resting on one arm or three arms), which indicated
that the substrate coupling and the collection geometry
played a role in the lasing spectra.
[153]
Intercavity coupling
between different arms was also identified as a possible
factor affecting the lasing threshold and the mode structure,
although this effect is likely not very significant since re-
moval of one tetrapod arm had only a small effect on the
lasing properties, as shown in Figure 14.
[153]
This is in agree-
ment with other reports which have shown that each leg of
the tetrapod acts as an individual laser.
[151, 152]
The lasing
from multiple tetrapod arms occurs only if all the arms are
excited, otherwise lasing will occur only from the excited
arm due to high losses in the non-excited tetrapod arms.
[152]
3.3. Factors Affecting the Achievement of Stimulated
Emission
It has been pointed out that very high lasing thresholds
are obtained in ZnO films with poor crystallinity.
[143]
In-
crease in the threshold with a decrease in crystal quality was
attributed to an increased concentration of nonradiative de-
fects.
[143]
On the other hand, lasing has been successfully ob-
tained from nanorods grown by hydrothermal meth-
ods,
[134, 154]
which typically have inferior crystallinity to sam-
ples fabricated by vapor deposition due to the low synthesis
temperature. The presence of strong defect emission does
not prevent stimulated emission, since the UV-to-visible
emission intensity ratio increases with increasing excitation
power.
[36]
Also, the long decay time of spontaneous emis-
sion, which indicates excellent crystal quality, is not necessa-
Figure 14. ZnO tetrapod manipulation and lasing in various configurations. a) Spectra recorded at
91, 284, 468, and 738 mJ cm
2
, showing stimulated emission in the three-arms-down configura-
tion. Inset (left): Power dependence curve showing a lasing threshold of 430 mJ cm
2
. Inset
(right): PL image of the lasing tetrapod depicting the vertical arm as a bright spot in the middle of
the structure. Scale bar =5 mm. b) Spectra recorded at 255, 454, 596, and 766 mJ cm
2
after flip-
ping the tetrapod into the three-arms-up configuration. The lasing threshold for this geometry
increased to 520 mJ cm
2
and the mode shape was drastically altered in comparison to (a).
Inset: PL image of the lasing tetrapod. Scale bar =5 mm. c) SEM image of the same tetrapod in
(a), (b), and (d) after removing one of the arms with a micromanipulator. Scale bar =1 mm.
d) Lasing spectra of the three-armed tetrapod shown in (c). The mode structure is similar to (b),
but the lasing threshold increases further to 584 mJ cm
2
. Inset: PL image of the lasing tripod
showing larger scattering loss from the left (top arm in the SEM image) damaged arm. Scale
bar =5 mm. Reprinted with permission.
[153]
954 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
ry for the achievement of lasing. Biexponential decay with
time constants of 70 and 350 ps was reported for ZnO nano-
wires with a lasing threshold of 40 kWcm
2
.
[4]
On the other
hand, highly faceted rods with long luminescence decay
times (116 ps and 1.2 ns) exhibited a lasing threshold of
45 mJcm
2
or 150 MWcm
2
.
[41]
It has been shown that stimulated emission can be ach-
ieved in nanostructures with different decay times and dif-
ferent defect emissions, but it could not be achieved if poor
crystal quality and high cavity losses occur simultaneous-
ly.
[170]
The lasing threshold will therefore be mainly depend-
ent on the dimensions of the nanostructure (see [Eq. (5)]),
quality of the cavity, and experimental conditions. Also, the
lasing thresholds obtained for different excitation wave-
lengths should not be directly compared due to different ab-
sorption of ZnO at different wavelengths. Due to great var-
iations in the lasing thresholds of individual nanowires
[138]
and different experimental conditions, it is difficult to com-
pare the lasing thresholds from different reports in the liter-
ature. Sometimes the reported results are contradictory. For
example, it was reported that nanoribbons had a higher
threshold compared to tetrapods and nanowires,
[153]
while
nanoribbon/nanocomb mixtures synthesized by a different
method exhibited a lower threshold than tetrapods and
nanorods.
[154]
However, the fact that lasing was demonstrat-
ed in a great variety of nanostructure morphologies indi-
cates that optically excited stimulated emission is easy to
achieve in the majority of ZnO nanostructures. Improved
crystallinity and larger dimensions can result in lower lasing
thresholds, though some variation among nanostructures
with similar dimensions fabricated in the same deposition
procedure is expected due to different conditions of the
cavity.
3.4. Time-Resolved Studies of Stimulated Emission
Time-resolved studies of stimulated emission in ZnO
have been performed by several
groups.
[41, 98, 127, 138141, 151, 152, 154, 163, 168]
The studies have been per-
formed on ZnO thin films,
[127, 140, 141]
nanowires,
[138, 139]
nano-
ribbons,
[139]
highly faceted rods,
[41]
nanoneedles,
[98]
nanorib-
bons/combs,
[154]
tetrapods,
[151, 152, 154, 168]
and nanorods.
[154, 163]
Typically, the stimulated emission decay time is much faster
than that of spontaneous emission, so that it may be below
the detection limit of some time-resolved photolumines-
cence systems.
[149]
Emissions in the EE and EHP regime ex-
hibit different behaviors with time.
[41, 98, 154, 168]
The compari-
son between the decay curves of the spontaneous emission,
EE, and EHP emissions from highly faceted rods is shown
in Figure 15. It is clear that although both types of stimulat-
ed emission have a shorter decay time compared to sponta-
neous emission, there are obvious differences in their tem-
poral evolution. EHP emission typically has a short rise
time (12 ps), which can be attributed to the thermalization
of the hot carriers.
[140, 141]
The EHP emission peak exhibits
some shifting with time, which was established by direct
measurements of the lasing spectra
[98, 140, 154, 168]
as well as by
measuring transient profiles of the lasing dynamics as a
function of wavelength.
[152]
The decay time of EHP emission
is typically just a few picoseconds.
[98, 151, 152, 154, 168]
It was sug-
gested that long cavity length, lower losses at end facets,
and lower defect concentrations would result in longer
decay times of the lasing.
[152]
Unlike EHP emission, stimulated emission in the EE
regime can exhibit a longer delay time before the onset of
the emission.
[41, 139, 154, 168]
This was attributed to the longer
time needed to achieve a high concentration of excitons in
the excited state.
[139]
Similar to EHP emission, decay time is
also typically just a few picoseconds.
[41, 154, 170]
With respect to
the evolution of the lasing spectra and any peak shifts in the
EE regime, some spectral shifts of the peaks can be ob-
served with time,
[154]
but it is difficult to analyze the data be-
cause the measurements have been performed on an ensem-
ble of the nanostructures. It should also be noted that as a
consequence of fast decay time of EHP emission and the
longer delay time of EE emission, coexistence of the two
can sometimes be observed in the time-resolved spectra ob-
tained at different times.
[154]
4. Nonlinear Optical Properties
As a consequence of its non-centrosymmetric crystal
structure, ZnO is expected to have nonzero second-order
susceptibility. Nonlinear properties have been studied for
different forms of ZnO.
[171189]
Nonlinear optical response of
C excitons has been measured by the four-wave mixing tech-
nique on ZnO single-crystal samples.
[174]
Second-harmonic
generation (SHG) was measured in ZnO single crystals,
[177]
thin films,
[171174, 176, 178185]
nanowires,
[139, 186]
and nanorib-
bons.
[139]
SHG in ZnO thin films is dependent on the deposi-
tion conditions,
[178, 180]
film thickness,
[171, 181]
crystalline struc-
ture,
[171, 179, 181]
orientation of the crystallites,
[182]
and the grain
shape.
[184]
The significant part of the SHG signal was found
to be generated at grain boundaries and interfaces.
[171]
It
was proposed that the film thickness more significantly af-
fects the second-order susceptibilities than the deposition
technique used.
[183]
Enhancement of the nonlinear suscepti-
Figure 15. Decay curves for three different emission regimes. The
inset shows an enlarged region in the range from 1 to 30 ps.
Reprinted from Ref. [41] .
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 955
Optical Properties of ZnO Nanostructures
bility compared to the bulk values was obtained for very
thin films.
[172]
The reduction of the second-order susceptibili-
ty with increasing film thickness was attributed to the
change in orientation of the polar axis as film thickness in-
creases.
[183]
It was also proposed that under certain experi-
mental conditions, third-harmonic generation (THG) com-
parable to a conventional SHG signal can be observed in
ZnO thin films.
[186]
Sputtered ZnO films exhibited both
second- and third-order nonlinear properties in spite of the
lack of a preferential growth direction, although the second-
order nonlinearities were lower than the bulk values.
[173]
However, THG has been less frequently studied
[173, 174, 186]
than SHG in ZnO.
While a transient SHG technique was used to study car-
rier dynamics in ZnO nanoribons and nanowires,
[139]
and
SHG and THG signals were measured in ZnO nano-
wires,
[186]
nonlinear optical properties of other ZnO nano-
structures have not been studied. In addition to SHG and
THG studies, two-photon
[187, 188]
and three-photon
[187]
spec-
troscopy and measurements of nonlinear refraction and ab-
sorption
[188]
of ZnO were also reported. Two-photon-in-
duced photoluminescence was also reported in ZnO micro-
tubes,
[189]
and two- and three-photon-induced luminescence
was observed in single-crystalline ZnO.
[175]
However, multi-
photon spectroscopy studies, as well as characterization of
the nonlinear optical properties of nanostructured ZnO in
general, have been scarce since both the measurement and
the theory are more complex compared to the characteriza-
tion of linear optical properties. Considering the variety of
available morphologies and possible fabrication methods for
ZnO nanostructures, it would be of particular interest if
more studies of the nonlinear optical properties of different
ZnO nanostructures were conducted.
5. Optical Properties of Doped ZnO
The optical properties of doped ZnO have been widely
studied.
[190228]
In general, the effect of doping on the optical
properties can be studied either by examining low-tempera-
ture UV spectra for evidence of the appearance of bound
exciton peaks not observed in undoped samples, or by ex-
amining the visible emission for changes in the defect emis-
sion spectra. The doping of ZnO is a research topic of con-
siderable interest in its own right, but the discussion will be
limited here to the actual doping of nanostructures and
nanocrystalline films. There are four main topics of interest
in doping ZnO: 1) doping with donor impurities to achieve
high n-type conductivity, 2) doping with acceptor impurities
to achieve p-type conductivity, 3) doping with rare-earth ele-
ments to achieve desired optical properties, and 4) doping
with transition metals to achieve desired magnetic proper-
ties.
While doping to achieve n-type conductivity is straight-
forward, the achievement of p-type conductivity is difficult
due to the presence of native defects. In addition to study-
ing the effects of impurities on optical, electrical, and mag-
netic properties, the effects of different impurities on the
orientation of ZnO nanorods fabricated by a combination
of chemical vapor deposition and laser ablation were also
studied.
[199]
It was found that some impurities (such as Er or
Mn) can result in preferential orientation of the nanorods
perpendicular to the substrate.
[199]
Doping is expected to
induce some changes in the morphology of the nanostruc-
tures, although the effects have not been systematically in-
vestigated for the majority of dopants and growth methods.
In addition to intentional doping, fabrication of hierarchical
ZnO structures where other elements are present in the
source material may result in the presence of secondary
phases and the incorporation of impurities in ZnO, as in the
case of Bi
2
O
3
-containing ZnO hierarchical nanostruc-
tures.
[192]
Group III and group IV elements are typically used to
dope ZnO with donor impurities. Halogen atoms can also
serve as donors in ZnO, and have the additional benefit of
reducing oxygen adsorption on surfaces,
[206]
but they are less
commonly used than group III dopants. Al-doped ZnO
(AZO) films are commonly proposed as a transparent con-
ductive oxide, which can replace for indium tin oxide elec-
trodes in organic optoelectronic devices. AZO nanosheets
and nanowalls have been reported.
[201]
It was found that the
addition of Al
2
O
3
to the source material resulted in Al
doping, but the morphology changed from nanowires to
nanosheets and nanowalls, while low-temperature cathodo-
luminescence spectra exhibited narrow donor bound-exciton
lines, with the I
6/8
line attributed to the Al donor,
[201]
in
agreement with other reported assignments of this donor
bound-exciton line.
[76]
Single-crystalline AlZnO nanowires/
nanotubes were also reported, and it was found that incor-
poration of Al resulted in an increased bandgap from
3.29 eV to 3.34 eV.
[194]
For the case of In doping, nanocrystalline films,
[225]
nano-
rods,
[198]
nanobelts,
[222]
and nanowires
[210, 216]
have been re-
ported. ZnO and ZnO:In nanorods were fabricated from
the reaction of zinc nitrate hydrate with hexamethylenetetr-
amine (for In doping, indium chloride was added).
[198]
No
significant differences in the morphology of the nanorods
were observed after doping. A blue shift of the absorption
peak and UV emission peak was observed, and the ratio of
UV-to-green emission decreased after doping.
[198]
On the
other hand, broadening and a red shift of the UV emission
peak was observed in In:ZnO nanobelts, which was attribut-
ed to a significant increase in the carrier density.
[222]
A simi-
lar red shift of the UV emission peak due to heavy In
doping was also observed in ZnO:In nanowires with a su-
perlattice structure.
[216]
In general doping with different donors produces broad-
ening of the UV emission peak, but the peak shift is de-
pendent on the dopant.
[210]
It was found that Sn doping pro-
duces the largest red shift of the UV emission, as well as the
appearance of strong green emission in doped ZnO nano-
wires.
[210]
On the other hand, another study of optical prop-
erties of Sn-doped ZnO nanowires reported no UV emission
shift (380 nm) and the appearance of new emission peaks at
396, 461, and 502 nm.
[219]
Obviously, since both undoped and
doped ZnO can exhibit different optical properties depend-
ent on the fabrication conditions, it is difficult to establish
how the properties will change after doping. The appear-
956 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
ance of new luminescence peaks is expected, but in the case
of significant increase in the carrier density, a red shift of
the near band-edge emission is also expected. It was also re-
ported that the optical properties of Sn-doped ZnO nano-
belts were dependent on the type of flowing gas used, illus-
trating the importance of the fabrication conditions.
[208]
In
addition to Sn, Pb represents another possible donor
dopant,
[219]
and Pb-doped ZnO nanowires have been report-
ed.
[207]
Pb doping was found to result in a red shift of the
emission peak with the peak position dependent on Pb con-
tent.
[207]
Sc doping also resulted in red-shifting of the PL
spectra with increasing concentration, remarkably similar to
the results observed for Pb-doped ZnO nanowires.
[213]
Acceptor dopants in ZnO are usually group V elements,
such as N, As, and P. Nitrogen doping was reported in ZnO
crystals,
[191]
nanocrystals,
[215]
and nanorods.
[204]
N doping of
ZnO crystals was found to result in the appearance of nitro-
gen-associated donoracceptor emissions in low-tempera-
ture PL spectra.
[191]
While N-doped nanocrystals showed
violet luminescence at room temperature (attributed to
defect emission),
[215]
N-doped nanorods showed a strong UV
emission at 3.31 eV and negligible defect emission.
[209]
In the
case of As-doped ZnO nanowires, unusual behavior in tem-
perature-dependent PL spectra has been observed, with ac-
ceptor bound-exciton emission detectable at room tempera-
ture.
[203, 214]
On the other hand, p-type P-doped ZnO films
exhibited more normal behavior with the acceptor bound-
exciton peak dominant at low temperatures and free-exciton
emission at room temperature.
[190]
ZnO nanostructures have also been doped with different
rare-earth elements such as Tb,
[228, 229]
Ce,
[217]
Eu,
[200, 204]
and
Dy.
[193]
In the case of Tb-doped ZnO nanoparticles, emission
from both Tb and surface states was observed.
[229]
Tb emis-
sion increased with increasing Tb concentration while the
emission from surface states decreased.
[228]
Ce incorporation
into 1D ZnO nanostructures results in the appearance of a
violet-blue emission and the disappearance of any green
defect emission.
[217]
Eu-related emission was observed from
ZnO:Eu nanorods for a suitable chosen excitation wave-
length.
[200, 204]
On the other hand, Dy-doped ZnO nanowires
exhibited only a UV emission of ZnO with only a very weak
emission attributed to Dy.
[193]
Therefore, when doping with
rare-earth elements, it is possible that their emission will be
masked by ZnO defect emission, and thus the excitation
wavelength must be carefully chosen to establish the effects
of doping on optical properties.
Doping with transition metals is expected to result in
changes in the magnetic properties of the material, and Mn-
doped ZnO has been predicted to be ferromagnetic at room
temperature. Therefore, there has been considerable interest
in the fabrication and characterization of transition-metal-
doped ZnO nanostructures. Mn-doped nanocrystalline
films,
[227]
tubes,
[195]
nanorods,
[197]
multileg nanostructures,
[211]
nanobelts,
[224]
and tetrapods
[223]
have been reported. Other
dopants include Ni (nanowire arrays)
[196]
and Co (nanoclus-
ter films).
[202]
Mn-doped ZnO rods were found to be ferro-
magnetic at room temperature,
[197]
but both the magnetic
and optical properties of the Mn-doped nanostructures are
strongly dependent on the fabrication conditions. Mn
doping was found to quench green emission,
[227]
although
other studies reported reduction in both UV and defect
emission.
[195]
Decrease in UV emission and the appearance
of green emission after Mn incorporation have also been re-
ported.
[197]
In addition, a blue shift and increase in intensity
of the UV peak were found after Mn doping.
[211]
Very simi-
lar spectra of ZnO and Mn-implanted ZnO were observed
after annealing an implanted sample at 8008C.
[224]
A similar
UV-to-green emission ratio has been observed in undoped
and Mn-doped ZnO tetrapods.
[223]
Obviously, the change in
the optical properties is strongly dependent on the method
of incorporation of Mn, fabrication conditions, and proper-
ties of undoped ZnO fabricated under similar conditions. In
the case of Ni doping, a red shift of the UV emission with
no significant change in the visible part of the spectrum was
observed.
[196]
Co doping also resulted in a small red shift of
the UV peak, as well as peak broadening.
[202]
Other dopants whose effects on the optical properties of
ZnO nanostructures have been studied include
sulfur
[205, 212, 221, 226]
and copper.
[218, 220]
Enhancement of green
emission with S doping has been reported,
[205, 221]
as well as
the change in shape of the broad green defect emission.
[212]
Either no significant shift
[221]
or, in contrast, a blue shift of
the UV emission peak
[205, 212, 226]
has been reported. In case of
Cu doping, broad PL spectra extending from the UV to the
red spectral region were observed in Cu:ZnO nanowires.
[220]
However, Cu-doped ZnO nanowires prepared by a different
method only showed an increased red shift of the emission
peak with increasing copper content.
[218]
Therefore, it can be
concluded that regardless of the type of dopant, the optical
properties of the nanostructures have a very strong depend-
ence on fabrication conditions. Thus, very careful interpreta-
tion of the measured spectra of doped ZnO nanostructures
is necessary.
6. Conclusions and Outlook
A great variety of ZnO nanostructures have been re-
ported. Their optical properties have been studied mostly at
room temperature, although variable-temperature photolu-
minescence studies on different ZnO nanostructures have
now been reported. Similar to other forms of ZnO, the
origin of different defect luminescence peaks remains unre-
solved. In general, the optical properties of various nano-
structures are very similar to those reported for thin films,
and the observed size effects in nanorods and nanowires are
the result of different surface-to-volume ratios rather than
through quantum confinement. All of the defect emissions
reported in nanostructures can also be found in thin-film or
bulk ZnO samples. The least-controversial defect emission
is the broad yellow luminescence commonly observed in
samples prepared from aqueous solutions, which likely origi-
nates from transitions involving interstitial oxygen; the
origin of other defect emissions require further study.
Although several methods for reducing or eliminating
defect emissions, such as annealing in an Ar/H
2
mixture for
yellow emission and surface functionalization and hydrogen
treatments for green emission, comprehensive study on the
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 957
Optical Properties of ZnO Nanostructures
relationship between fabrication conditions and visible
emission spectra is still needed. In order to conclusively
identify the nature of defects, it will likely be necessary to
combine PL studies with other experimental techniques
such as EPR, carrier-concentration determination, and X-
ray photoelectron spectroscopy (XPS). In particular, anneal-
ing studies in different atmospheres and at different temper-
atures may be a useful tool in establishing the origin of the
defect emission since annealing may result in the change of
optical properties without changing the morphology.
As for the stimulated emission, while ultrafast carrier
dynamics has been well studied in the EHP regime, further
studies are needed for lasing due to excitonexciton scatter-
ing, which is of higher practical interest. Also, for practical
applications of ZnO lasers it is necessary to achieve electri-
cally pumped lasing in ZnO. Closely related to this issue is
the achievement of reliable p-type doping with high carrier
concentrations and mobilities, which is difficult due to the
presence of native defects in ZnO. Although several groups
have reported p-type doping in ZnO, this issue is still under
scrutiny.
[1]
Research on doped ZnO nanostructures has been
scarce compared to research on the doping of thin films and
on undoped nanostructures. Finally, while there are numer-
ous reports on the linear optical properties of ZnO nano-
structures, much work remains to be done on studying their
nonlinear properties.
Acknowledgements
This work is partly supported by the Research Grant Council
of the Hong Kong Special Administrative Region, China (HKU
7019/04P) and a University Development Fund grant of the
University of Hong Kong.
[1] . zgr, Ya. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S.
Dogan, V. Avrutin, S.-J. Cho, H. Morko, J. Appl. Phys. 2005,
98, 041301.
[2] R. Triboulet, J. Perriere, Prog. Cryst. Growth Charact. Mater.
2003, 47, 65.
[3] R. Janisch, P. Gopal, N. A. Spaldin, J. Phys. Condens. Matter
2005, 17, R657.
[4] M. H. Huang, S. Mao, H. Feick, H. Yan, Y. Wu, H. Kind, E. Weber,
R. Russo, P. Yang, Science 2001, 292, 1897.
[5] M. H. Huang, Y. Wu, H. Feick, N. Tran, E. Weber, P. Yang, Adv.
Mater. 2001, 13, 113.
[6] B. D. Yao, Y. F. Chan, N. Wang, Appl. Phys. Lett. 2002, 81, 757.
[7] L. E. Greene, M. Law, D. H. Tan, M. Montano, J. Goldberger, G.
Somorjai, P. Yang, Nano Lett. 2005, 5, 1231.
[8] C. Liu, J. A. Zapien, Y. Yao, X. Meng, C. S. Lee, S. Fan, Y. Lifshitz,
S. T. Lee, Adv. Mater. 2003, 15, 838.
[9] S. Y. Li, P. Lin, C. Y. Lee, T. Y. Tseng, J. Appl. Phys. 2004, 95,
3711.
[10] B. Liu, H. C. Zeng, J. Am. Chem. Soc. 2003, 125, 4430.
[11] M. Guo, P. Diao, S. Cai, J. Solid State Chem. 2005, 178, 1864.
[12] W. I. Park, Y. H. Jun, S. W. Jung, G.-C. Yi, Appl. Phys. Lett. 2003,
82, 964.
[13] A. B. Hartanto, X. Ning, Y. Nakata, T. Okada, Appl. Phys. A
2003, 78, 299.
[14] W. D. Yu, X. M. Li, X. D. Gao, Appl. Phys. Lett. 2004, 84, 2658.
[15] Y. Dai, Y. Zhang, Q. K. Li, C. W. Nan, Chem. Phys. Lett. 2002,
358, 83.
[16] Y. Dai, Y. Zhang, Z. L. Wang, Solid State Commun. 2003, 126,
629.
[17] V. A. L. Roy, A. B. Djurisic, W. K. Chan, J. Gao, H. F. Lui, C.
Surya, Appl. Phys. Lett. 2003, 83, 141.
[18] H. Yan, R. He, J. Pham, P. Yang, Adv. Mater. 2003, 15, 402.
[19] Z. W. Pan, Z. R. Dai, Z. L. Wang, Science 2001, 291, 1947.
[20] H. Yan, J. Johnson, M. Law, R. He, K. Knutsen, J. R. McKinney, J.
Pham, R. Saykally, P. Yang, Adv. Mater. 2003, 15, 1907.
[21] Y. B. Li, Y. Bando, T. Sato, K. Kurashima, Appl. Phys. Lett.
2002, 81, 144.
[22] J. Y. Lao, J. G. Wen, Z. F. Ren, Nano Lett. 2002, 2, 1287.
[23] J. Y. Lao, J. Y. Huang, D. Z. Wang, Z. F. Ren, Nano Lett. 2003, 3,
235.
[24] Y. J. Xing, Z. H. Xi, X. D. Zhang, J. H. Song, R. M. Wang, J. Xu,
Z. Q. Xue, D. P. Yu, Solid State Commun. 2004, 129, 671.
[25] J.-H. Park, H.-J. Choi, Y.-J. Choi, S.-H. Sohn, J.-G. Park, J. Mater.
Chem. 2004, 14, 35.
[26] Z. L. Wang, X. Y. Kong, Y. Ding, P. Gao, W. L. Hughes, R. Yang, Y.
Zhang, Adv. Funct. Mater. 2004, 14, 943.
[27] P. X. Gao, Z. L. Wang, Appl. Phys. Lett. 2004, 84, 2883.
[28] X. Y. Kong, Z. L. Wang, Nano Lett. 2003, 3, 1625.
[29] Z. Y. Fan, J. G. Lu, J. Nanosci. Nanotechnol. 2005, 5, 1561.
[30] G. C. Yi, C. R. Wang, W. I. Park, Semicond. Sci. Technol. 2005,
20, S22.
[31] Y. W. Heo, D. P. Norton, L. C. Tien, Y. Kwon, B. S. Kang, F. Ren,
S. J. Pearton, J. R. LaRoche, Mater. Sci. Eng. R 2004, 47, 1.
[32] J. Grabowska, A. Meaney, K. K. Nanda, J.-P. Mosnier, M. O.
Henry, J.-R. Duclre, E. McGlynn, Phys. Rev. B 2005, 71,
115439.
[33] S. Chen, Y. Liu, C. Shao, R. Mu, Y. Lu, J. Zhang, D. Shen, X. Fan,
Adv. Mater. 2005, 17, 586.
[34] H.-C. Hsu, W.-F. Hsieh, Solid State Commun. 2004, 131, 371.
[35] B. P. Zhang, N. T. Binh, Y. Segawa, K. Wakatsuki, N. Usami,
Appl. Phys. Lett. 2003, 83, 1635.
[36] A. B. Djurisic, W. C. H. Choy, V. A. L. Roy, Y. H. Leung, C. Y.
Kwong, K. W. Cheah, T. K. Gundu Rao, W. K. Chan, H. F. Lui, C.
Surya, Adv. Funct. Mater. 2004, 14, 856.
[37] W. I. Park, Y. H. Jun, S. W. Jung, G.-C. Yi, Appl. Phys. Lett. 2003,
82, 964.
[38] B. P. Zhang, C. Y. Liu, Y. Segawa, Y. Kashiwaba, K. Haga, Thin
Solid Films 2005, 474, 165.
[39] J. Jie, G. Wang, Y. Chen, X. Han, Q. Wang, B. Xu, J. G. Hou, Appl.
Phys. Lett. 2005, 86, 031909.
[40] H.-W. Suh, G.-Y. Kim, Y.-S. Jung, W.-K. Choi, D. Byun, J. Appl.
Phys. 2005, 97, 044305.
[41] W. M. Kwok, A. B. Djurisic, Y. H. Leung, W. K. Chan, D. L. Phil-
lips, H. Y. Chen, C. L. Wu, S. Gwo, M. H. Xie, Chem. Phys. Lett.
2005, 412, 141.
[42] Y. H. Tong, Y. C. Liu, S. X. Lu, L. Dong, S. J. Chen, Z. Y. Xiao, J.
Sol-Gel Sci. Technol. 2004, 30, 157.
[43] S. Ozaki, T. Tsuchiya, Y. Inokuchi, S. Adachi, Phys. Stat. Sol. A
2005, 202, 1325.
[44] X. H. Zhang, Y. C. Liu, X. H. Wang, S. J. Chen, G. R. Wang, J. Y.
Zhang, Y. M. Lu, D. Z. Shen, X. W. Fan, J. Phys. Condens. Matter
2005, 17, 3035.
[45] H.-C. Hsu, W.-F. Hsieh, Solid State Commun. 2004, 131, 371.
[46] L. E. Greene, M. Law, J. Goldberger, F. Kim, J. C. Johnson, Y.
Zhang, R. J. Saykally, P. Yang, Angew. Chem. 2003, 115, 3030;
Angew. Chem. Int. Ed. 2003, 42, 3031.
[47] X. Q. Meng, D. Z. Shen, J. Y. Zhang, D. X. Zhao, Y. M. Lu, L.
Dong, Z. Z. Zhang, Y. C. Liu, X. W. Fan, Solid State Commun.
2005, 135, 179.
[48] Y. Q. Chen, J. Jiang, Z. Y. He, Y. Su, D. Cai, L. Chen, Mater. Lett.
2005, 59, 3280.
958 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
[49] R.-C. Wang, C.-P. Liu, J.-L. Huang, S.-J. Chen, Appl. Phys. Lett.
2005, 87, 053103.
[50] S. Choopun, N. Hongsith, S. Tanunchai, T. Chairuangsri, C.
Krua-in, S. Singkarat, T. Vilathong, P. Mangkorntong, N. Man-
gkorntong, J. Cryst. Growth 2005, 282, 365.
[51] H. Zhou, H. Alves, D. M. Hofmann, W. Kriegseis, B. K. Meyer, G.
Kaczmarczyk, A. Hoffmann, Appl. Phys. Lett. 2002, 80, 210.
[52] L. Huang, S. Wright, S. Yang, D. Shen, B. Gu, Y. Du, J. Phys.
Chem. B 2004, 108, 19901.
[53] H. J. Fan, R. Scholz, F. M. Kolb, M. Zacharias, Appl. Phys. Lett.
2004, 85, 4142.
[54] H. J. Fan, R. Scholz, M. Zacharias, U. Gsele, F. Bertram, D. For-
ster, J. Christen, Appl. Phys. Lett. 2005, 86, 023113.
[55] D. Zhao, C. Andreazza, P. Andreazza, J. Ma, Y. Liu, D. Shen,
Chem. Phys. Lett. 2004, 399, 522.
[56] Q. Yang, K. Tang, J. Zuo, Y. Qian, Appl. Phys. A 2004, 79, 1847.
[57] H. J. Fan, R. Scholz, F. M. Kolb, M. Zacharias, U. Gsele, F. Heyr-
oth, C. Eisenschmidt, T. Hempel, J. Christen, Appl. Phys. A
2004, 79, 1895.
[58] F. Wang, Z. Ye, D. Ma, L. Zhu, F. Zhuge, J. Cryst. Growth 2005,
274, 447.
[59] W. Yu, X. Li, X. Gao, Cryst. Growth Des. 2005, 5, 151.
[60] R. Wu, Y. Yang, S. Cong, Z. Wu, C. Xie, H. Usui, K. Kawaguchi,
N. Koshizaki, Chem. Phys. Lett. 2005, 406, 457.
[61] S. M. Abrarov, Sh. U. Yuldashev, T. W. Kim, S. B. Lee, Y. H.
Kwon, T. W. Kang, Opt. Commun. 2005, 250, 111.
[62] F. Wen, W. Li, J.-H. Moon, J.-H. Kim, Solid State Commun. 2005,
135, 34.
[63] L. Weng, X. Zhang, S. Zhao, G. Zhou, Y. Zhou, J. Qi, Appl. Phys.
Lett. 2005, 86, 024108.
[64] H. T. Ng, B. Chen, J. Li, J. Han, M. Meyyappan, J. Wu, S. X. Li,
E. E. Haller, Appl. Phys. Lett. 2003, 82, 2023.
[65] C. X. Xu, X. W. Sun, X. H. Zhang, L. Ke, S. J. Chua, Nanotechnol-
ogy 2004, 15, 856.
[66] T. Hirai, Y. Asada, J. Coll. Interf. Science 2005, 284, 184.
[67] X. Liu, X. Wu, H. Cao, R. P. H. Chang, J. Appl. Phys. 2004, 95,
3141.
[68] Z. Chen, N. Wu, Z. Shan, M. Zhao, S. Li, C. B. Jiang, M. K. Chyu,
S. X. Mao, Scr. Mater. 2005, 52, 63.
[69] T. B. Hur, Y.-H. Hwang, H.-K. Kim, Appl. Phys. Lett. 2005, 86,
193113.
[70] Y. Gu, I. L. Kuskovsky, M. Yin, S. OBrien, G. F. Neumark, Appl.
Phys. Lett. 2004, 85, 3833.
[71] S. Hong, T. Joo, W. I. Park, Y. H. Jun, G.-C. Yi, Appl. Phys. Lett.
2003, 83, 4157.
[72] N. E. Hsu, W. K. Hung, Y. F. Chen, Appl. Phys. Lett. 2004, 96,
4671.
[73] I. Shalish, H. Temkin, V. Narayanamurti, Phys. Rev. B 2004, 69,
245401.
[74] W. S. Shi, B. Cheng, L. Zhang, E. T. Samulski, J. Appl. Phys.
2005, 98, 083502.
[75] C. Ye, X. Fang, Y. Hao, X. Teng, L. Zhang, J. Phys. Chem. B
2005, 109, 19758.
[76] B. K. Meyer, H. Alves, D. M. Hofmann, W. Kreigseis, D. Forster,
F. Bertram, J. Christen, A. Hoffmann, M. Strassburg, M. Dwor-
zak, U. Haboeck, A. V. Rodina, Phys. Stat. Sol. B 2004, 241,
231.
[77] A. Teke, . zgr, S. Dogan, X. Gu, H. Morko, B. Nemeth, J.
Nause, H. O. Everitt, Phys. Rev. B 2004, 70, 195207.
[78] J. Gutowski, N. Presser, I. Broser, Phys. Rev. B 1988, 38, 9746.
[79] M. Strassburg, A. Rodina, M. Dworzak, U. Haboeck, I. L. Krest-
nikov, A. Hoffmann, O. Gelhausen, M. R. Phillips, H. R. Alves,
A. Zeuner, D. M. Hofmann, B. K. Meyer, Phys. Stat. Sol. B 2004,
241, 607.
[80] D. C. Look, C. Cos kun, B. Claflin, G. C. Farlow, Physica B 2003,
340342, 32.
[81] D. C. Look, R. L. Jones, J. R. Sizelove, N. Y. Garces, N. C. Giles,
L. E. Halliburton, Phys. Stat. Sol. A 2003, 195, 171.
[82] K. Thonke, T. Gruber, N. Teofoliv, R. Schnfelder, A. Waag, R.
Sauer, Physica B 2001, 308310, 945.
[83] H. Alves, D. Pfisterer, A. Zeuner, T. Riemann, J. Christen, D. M.
Hofmann, B. K. Meyer, Opt. Mater. 2003, 23, 33.
[84] L. Wang, N. C. Giles, J. Appl. Phys. 2003, 94, 973.
[85] B. Lin, Z. Fu, Y. Jia, Appl. Phys. Lett. 2001, 79, 943.
[86] P. S. Xu, Y. M. Sun, C. S. Shi, F. Q. Xu, H. B. Pan, Nucl. Instrum.
Methods Phys. Res. B 2003, 199, 286.
[87] S. A. M. Lima, F. A. Sigoli, M. Jafelicci Jr. , M. R. Davolos, Inter-
net Symp. Food Allergens Intern. J. Inorg. Mat. 2001, 3, 749.
[88] N. Y. Garces, L. Wang, L. Bai, N. C. Giles, L. E. Halliburton, G.
Cantwell, Appl. Phys. Lett. 2002, 81, 622.
[89] D. C. Reynolds, D. C. Look, B. Jogai, J. Appl. Phys. 2001, 89,
6189.
[90] A. van Dijken, E. Meulenkamp, D. Vanmaekelbergh, A. Meijer-
ink, J. Phys. Chem. B 2000, 104, 1715.
[91] A. van Dijken, E. Meulenkamp, D. Vanmaekelbergh, A. Meijer-
ink, J. Lumin. 2000, 90, 123.
[92] Y. W. Heo, D. P. Norton, S. J. Pearton, J. Appl. Phys. 2005, 98,
073502.
[93] Q. X. Zhao, P. Klason, M. Willander, H. M. Zhong, W. Lu, J. H.
Yang, Appl. Phys. Lett. 2005, 87, 211912.
[94] K. Vanheusden, C. H. Seager, W. L. Warren, D. R. Tallant, J. A.
Voigt, Appl. Phys. Lett. 1996, 68, 403.
[95] Y. G. Wang, S. P. Lau, X. H. Zhang, H. W. Lee, S. F. Yu, B. K. Tay,
H. H. Hng, Chem. Phys. Lett. 2003, 375, 113.
[96] D. Li, Y. H. Leung, A. B. Djurisic, Z. T. Liu, M. H. Xie, S. L. Shi,
S. J. Xu, W. K. Chan, Appl. Phys. Lett. 2004, 85, 1601.
[97] R. B. M. Cross, M. M. De Souza, E. M. Sankara Narayanan,
Nanotechnology 2005, 16, 2188.
[98] W. M. Kwok, Y. H. Leung, A. B. Djurisic, W. K. Chan, D. L. Phil-
lips, Appl. Phys. Lett. 2005, 87, 093108.
[99] S. A. Studenikin, N. Golego, M. Cocivera, J. Appl. Phys. 1998,
84, 2287.
[100] H. C. Ong, G. T. Du, J. Cryst. Growth 2004, 265, 471.
[101] R. B. Lauer, J. Phys. Chem. Solids 1973, 34, 249.
[102] M. Gomi, N. Oohira, K. Ozaki, M. Koyano, Jpn. J. Appl. Phys.
2003, 42, 481.
[103] C. C. Lin, H. P. Chen, H. C. Liao, S. Y. Chen, Appl. Phys. Lett.
2005, 86, 183103.
[104] A. B. Djurisic, Y. H. Leung, W. C. H. Choy, K. W. Cheah, W. K.
Chan, Appl. Phys. Lett. 2004, 84, 2635.
[105] M. Liu, A. H. Kitai, P. Mascher, J. Lumin. 1992, 54, 35.
[106] T. Koida, S. Chichibu, A. Uedono, A. Tsukazaki, M. Kawasaki, T.
Sota, Y. Segawa, H. Koinuma, Appl. Phys. Lett. 2003, 82, 532.
[107] F. D. Auret, S. A. Goodman, M. J. Legodi, W. E. Meyer, D. C.
Look, Appl. Phys. Lett. 2002, 80, 1340.
[108] H. J. Ko, Y. F. Chen, Z. Zhu, T. Yao, I. Kobayashi, H. Uchiki, Appl.
Phys. Lett. 2000, 76, 1905.
[109] H. J. Ko, Y. F. Chen, T. Yao, K. Miyajima, A. Yamamoto, T. Goto,
Appl. Phys. Lett. 2000, 77, 537.
[110] D. W. Hamby, D. A. Lucca, M. J. Klopfstein, G. Cantwell, J. Appl.
Phys. 2003, 93, 3214.
[111] H. J. Ko, Y. Chen, S. K. Hong, T. Yao, J. Cryst. Growth 2000, 209,
816.
[112] S. W. Kim, S. Fujita, S. Fujita, Appl. Phys. Lett. 2005, 86,
153119.
[113] S. A. Studenikin, M. Cocivera, W. Kellner, H. Pascher, J. Lumin.
2000, 91, 223.
[114] B. P. Zhang, N. T. Binh, Y. Segawa, Y. Kashiwaba, K. Haga, Appl.
Phys. Lett. 2004, 84, 586.
[115] Y. Harada, H. Kondo, N. Ichimura, S. Hashimoto, J. Lumin.
2000, 8789, 405.
[116] Y. Kashiwaba, K. Haga, H. Watanabe, B. P. Zhang, Y. Segawa, K.
Wakatsuki, Phys. Stat. Sol. B 2002, 229, 921.
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 959
Optical Properties of ZnO Nanostructures
[117] M. Haupt, A. Ladenburger, R. Sauer, K. Thonke, R. Glass, W.
Roos, J. P. Spatz, H. Rauscher, S. Riethmller, M. Mller, J.
Appl. Phys. 2003, 93, 6252.
[118] H. Kato, M. Sano, K. Miyamoto, T. Yao, J. Cryst. Growth 2004,
265, 375.
[119] B.-P. Zhang, L.-H. Manh, K. Wakatsuki, T. Ohnishi, M. Lippmaa,
N. Usami, M. Kawasaki, Y. Segawa, Jpn, J. Appl. Phys. 2003,
42, 2291.
[120] Y. Harada, S. Hashimoto, Phys. Rev. B 2003, 68, 045421.
[121] X. Wang, S. Yang, J. Wang, M. Li, X. Jiang, G. Du, X. Liu, R. P. H.
Chang, Opt. Quantum Electron. 2002, 34, 883.
[122] A. Tsukazaki, A. Ohtomo, M. Kawasaki, T. Makino, C. H. Chia, Y.
Segawa, H. Koinuma, Appl. Phys. Lett. 2004, 84, 3858.
[123] C. G. Van de Walle, Physica B 2001, 308, 899.
[124] A. Kobayashi, O. F. Sankey, J. D. Dow, Phys. Rev. B 1983, 28,
946.
[125] W. Gpel, U. Lampe, Phys. Rev. B 1980, 22, 6447.
[126] W. Hirschwald, P. Bonasewicz, L. Ernzt, M. Grade, D. Hoffmann,
S. Krebs, R. Littbarski, G. Neumann, M. Grunze, D. Kolb, H. J.
Schulz in Current topics in materials science, vol. 7 (Ed: E.
Kaldis), North-Holland, Amsterdam 1981, Ch. 3.
[127] . zgr, A. Teke, C. Liu, S.-J. Cho, H. Morko, H. O. Everitt,
Appl. Phys. Lett. 2004, 84, 3223.
[128] H. Cao, Y. G. Zhao, H. C. Ong, S. T. Ho, J. Y. Dai, J. Y. Wu, R. P. H.
Chang, Appl. Phys. Lett. 1998, 73, 3656.
[129] X. W. Sun, S. F. Yu, C. X. Xu, C. Yuen, B. J. Chen, S. Li, Jpn. J.
Appl. Phys. 2003, 42, L1229.
[130] J.-H. Choy, E.-S. Jang, J.-H. Won, J.-H. Chung, D.-J. Jang, Y.-W.
Kim, Appl. Phys. Lett. 2004, 84, 287.
[131] Z. Qiu, K. S. Wong, M. Wu, W. Lin, H. Xu, Appl. Phys. Lett.
2004, 84, 2739.
[132] K. Govender, D. S. Boyle, P. OBrien, D. Binks, D. West, D. Cole-
man, Adv. Mater. 2002, 14, 1221.
[133] S. F. Yu, C. Yuen, S. P. Lau, W. I. Park, G.-C. Yi, Appl. Phys. Lett.
2004, 84, 3241.
[134] J.-H. Choy, E.-S. Jang, J.-H. Won, J.-H. Chung, D.-J. Jang, Y.-W.
Kim, Adv. Mater. 2003, 15, 1911.
[135] H. Yan, J. Johnson, M. Law, R. He, K. Knutsen, J. R. McKinney, J.
Pham, R. Saykally, P. Yang, Adv. Mater. 2003, 15, 1907.
[136] K. Bando, T. Sawabe, K. Asaka, Y. Matsumoto, J. Lumin. 2004,
108, 385.
[137] H. Yan, R. He, J. Johnson, M. Law, R. J. Saykally, P. Yang, J. Am.
Chem. Soc. 2003, 125, 4728.
[138] J. C. Johnson, H. Yan, P. Yang, R. J. Saykally, J. Phys. Chem. B
2003, 107, 8816.
[139] J. C. Johnson, K. P. Knutsen, H. Yan, M. Law, Y. Zhang, P. Yang,
R. J. Saykally, Nano Lett. 2004, 4, 197.
[140] J. Takeda, S. Kurita, Y. Chen, T. Yao, Int. J. Modern Phys. B
2001, 15, 3669.
[141] A. Yamamoto, T. Kido, T. Goto, Y. Chen, T. Yao, A. Kasuya, J.
Cryst. Growth 2000, 214, 308.
[142] X. Q. Zhang, I. Suemune, H. Kumano, J. Wang, S. H. Huang, J.
Appl. Phys. 2004, 96, 3733.
[143] X. Liu, A. Yamilov, X. Wu, J.-G. Zheng, H. Cao, R. P. H. Cheng,
Chem. Mater. 2004, 16, 5414.
[144] B. Guo, Z. Ye, K. S. Wong, J. Cryst. Growth 2003, 253, 252.
[145] H.-C. Hsu, C.-Y. Wu, W.-F. Hsieh, J. Appl. Phys. 2005, 97,
064315.
[146] Y. Segawa, A. Ohtomo, M. Kawasaki, H. Koinuma, Z. K. Tang, P.
Yu, G. K. L. Wong, Phys. Stat. Sol. B 1997, 202, 669.
[147] P. Yang, H. Yan, S. Mao, R. Russo, J. Johnson, R. Saykally, N.
Morris, J. Pham, R. He, H.-J. Choi, Adv. Funct. Mater. 2002, 12,
323.
[148] C. Liu, J. A. Zapien, Y. Yao, X. Meng, C. S. Lee, S. Fan, Y. Lifshitz,
S. T. Lee, Adv. Mater. 2003, 15, 838.
[149] L. Cao, B. Zou, C. Li, Z. Zhang, S. Xie, G. Yang, Europhys. Lett.
2004, 68, 740.
[150] C. Yuen, S. F. Yu, X. W. Sun, C. X. Xu, E. S. P. Leong, S. P. Lau,
C. K. Chen, Jpn. J. Appl. Phys. 2004, 43, 5273.
[151] J. M. Sarko, J. K. Song, C. W. Blackledge, I. Swart, S. R. Leone,
S. Li, Y. Zhao, Chem. Phys. Lett. 2005, 404, 171.
[152] J. K. Song, J. M. Szarko, S. R. Leone, S. Li, Y. Zhao, J. Phys.
Chem. B 2005, 109, 15749.
[153] D. J. Sirbuly, M. Law, H. Yan, P. Yang, J. Phys. Chem. B 2005,
109, 15190.
[154] A. B. Djurisic, W. M. Kwok, Y. H. Leung, W. K. Chan, D. L. Phil-
lips, J. Phys. Chem. B 2005, 109, 19228.
[155] A. Stassinopoulos, R. N. Das, E. P. Giannelis, S. H. Anastasia-
dis, D. Anglos, Appl. Surf. Sci. 2005, 247, 18.
[156] S. P. Lau, H. Y. Yang, S. F. Yu, H. D. Li, M. Tanemura, T. Okita, H.
Hatano, H. H. Hng, Appl. Phys. Lett. 2005, 87, 013104.
[157] H. D. Li, S. F. Yu, A. P. Abiyasa, C. Yuen, S. P. Lau, H. Y. Yang,
E. S. P. Leong, Appl. Phys. Lett. 2005, 86, 261111.
[158] S. F. Yu, C. Yuen, S. P. Lau, H. W. Lee, Appl. Phys. Lett. 2004,
84, 3244.
[159] C. Yuen, S. F. Yu, E. S. P. Leong, H. Y. Yang, S. P. Lau, N. S. Chen,
H. H. Hng, Appl. Phys. Lett. 2005, 86, 031112.
[160] S. P. Lau, H. Y. Yang, S. F. Yu, C. Yuen, E. S. P. Leong, H. D. Li,
H. H. Hng, Small 2005, 1, 956.
[161] C. X. Xu, X. W. Sun, C. Yuen, S. F. Yu, Z. L. Dong, Appl. Phys.
Lett. 2005, 86, 011118.
[162] H. Cao, Y. G. Zhao, S. T. Ho, E. W. Seelig, Q. H. Wang, R. P. H.
Chang, Phys. Rev. Lett. 1999, 82, 2278.
[163] X. Han, G. Wang, Q. Wang, L. Cao, R. Liu, B. Zou, J. G. Hou,
Appl. Phys. Lett. 2005, 86, 223106.
[164] Z. K. Tang, G. K. L. Wong, P. Yu, M. Kawasaki, A. Ohtomo, H. Koi-
numa, Y. Segawa, Appl. Phys. Lett. 1998, 72, 3270.
[165] P. Zu, Z. K. Tang, G. K. L. Wong, M. Kawasaki, A. Ohtomo, H.
Koinuma, Y. Segawa, Solid State Commun. 1997, 103, 459.
[166] T. Okada, K. Kawashima, M. Ueda, Appl. Phys. A 2005, 81,
907.
[167] Y. Sun, J. B. Ketterson, G. K. L. Wong, Appl. Phys. Lett. 2000,
77, 2322.
[168] Y. H. Leung, W. M. Kwok, A. B. Djurisic, D. L. Phillips, W. K.
Chan, Nanotechnology 2005, 16, 579.
[169] D. S. Wiersma, A. Lagendijk, Phys. Rev. E 1996, 54, 4256.
[170] W. M. Kwok, A. B. Djurisic, Y. H. Leung, W. K. Chan, D. L. Phil-
lips, Appl. Phys. Lett. 2005, 87, 223111.
[171] H. Cao, J. Y. Wu, H. C. Ong, J. Y. Dai, R. P. H. Chang, Appl. Phys.
Lett. 1998, 73, 572.
[172] X. Q. Zhang, Z. K. Tang, M. Kawasaki, A. Ohtomo, H. Koinuma,
Thin Solid Films 2004, 450, 320
[173] M. C. Larciprete, D. Haertle, A. Belardini, M. Bertolotti, F. Sarto,
P. Gnter, Appl. Phys. B 2006, 82, 431.
[174] K. Hazu, K. Torii, T. Sota, S. Adachi, S. F. Chichibu, G. Cantwell,
D. C. Reynolds, C. W. Litton, J. Appl. Phys. 2004, 95, 5498.
[175] D. C. Dai, S. J. Xu, S. L. Shi, M. H. Xie, C. M. Che, Opt. Lett.
2005, 30, 3377.
[176] G. I. Petrov, V. Shcheslavskiy, V. V. Yakovlev, I. Ozerov, E. Chel-
nokov, W. Marine, Appl. Phys. Lett. 2003, 83, 3993.
[177] G. Wang, G. K. L. Wong, J. B. Ketterson, Appl. Opt. 2001, 40,
5436.
[178] M. C. Larciprete, D. Passeri, F. Michelotti, S. Paolini, C. Sibilia,
M. Bertolotti, A. Beraldini, F. Sarto, F. Somma, S. Lo Mastro, J.
Appl. Phys. 2005, 97, 023501.
[179] X. Q. Zhang, Z. K. Tang, M. Kawasaki, A. Ohtomo, H. Koinuma,
J. Phys. Condens. Matter 2003, 15, 5191.
[180] C. Y. Liu, B. P. Zhang, N. T. Binh, Y. Segawa, Opt. Commun.
2004, 237, 65.
[181] X. Q. Zhang, Z. K. Tang, M. Kawasaki, A. Ohtomo, H. Koinuma,
Thin Solid Films 2004, 450, 320.
[182] U. Neumann, R. Grunwald, U. Griebner, G. Steinmeyer, M.
Schmidbauer, W. Seeber, Appl. Phys. Lett. 2005, 87, 171108.
960 www.small-journal.com 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim small 2006, 2, No. 8-9, 944 961
reviews A. B. Djurisic and Y. H. Leung
[183] G. Wang, G. T. Kiehne, G. K. L. Wong, J. B. Ketterson, X. Liu,
R. P. H. Chang, Appl. Phys. Lett. 2002, 80, 401.
[184] U. Neumann, R. Grunwald, U. Griebner, G. Steinmeyer, W.
Seeber, Appl. Phys. Lett. 2004, 84, 170.
[185] T. Tritschler, O. D. Mcke, M. Wegener, U. Morgner, F. X. Krt-
ner, Phys. Rev. Lett. 2003, 90, 217404.
[186] J. C. Johnson, H. Yan, R. D. Schaller, P. B. Peterson, P. Yang, R. J.
Saykally, Nano Lett. 2002, 2, 279.
[187] J. Wrzesinski, D. Frhlich, J. Cryst. Growth 1998, 185, 686.
[188] J.-H. Lin, Y.-J. Chen, H.-Y. Lin, W.-F. Hsieh, J. Appl. Phys. 2005,
97, 033526.
[189] C. F. Zhang, Z. W. Dong, G. J. You, S. X. Qian, H. Deng, H. Gao,
L. P. Yang, Y. Li, Appl. Phys. Lett. 2005, 87, 051920.
[190] F. X. Xiu, Z. Yang, L. J. Mandalapu, J. L. Liu, W. P. Beyermann,
Appl. Phys. Lett. 2006, 88, 052106.
[191] B. G. Wang, M. J. Callahan, L. O. Bouthillette, C. C. Xu, M. J.
Suscavage, J. Crystal Growth 2006, 287, 381.
[192] C. K. Xu, K. Rho, J. Chun, D. E. Kim, Nanotechnology 2006, 17,
60.
[193] G. S. Wu, Y. L. Zhuang, Z. Q. Lin, X. Y. Yuan, T. Xie, L. D. Zhang,
Physica E 2006, 31, 5.
[194] R. C. Wang, C. P. Liu, J. L. Huang, S. J. Chen, Appl. Phys. Lett.
2006, 88, 023111.
[195] C. X. Xu, X. W. Sun, Z. L. Dong, S. T. Tan, Y. P. Cui, B. P. Wang, J.
Appl. Phys. 2005, 98, 113513.
[196] H. He, C. S. Lao, L. J. Chen, D. Davidovic, Z. L. Wang, J. Am.
Chem. Soc. 2005, 127, 16376.
[197] J. M. Baik, J. L. Lee, Adv. Mater. 2005, 17, 2745.
[198] Y. W. Chen, Y. C. Liu, S. X. Lu, C. S. Xu, C. L. Shao, C. Wang, J. Y.
Zhang, Y. M. Lu, D. Z. Shen, X. W. Fan, J. Chem. Phys. 2005,
123, 134701.
[199] T. Hirate, S. Sasaki, W. C. Li, H. Miyashita, T. Kimpara, T.
Satoh, Thin Solid Films 2005, 487, 35.
[200] A. Ishizumi, Y. Taguchi, A. Yamamoto, Y. Kanemitsu, Thin Solid
Films 2005, 486, 50.
[201] A. Rahm, G. W. Yang, M. Lorenz, T. Nobis, J. Lenzner, G.
Wagner, M. Grundmann, Thin Solid Films 2005, 486, 191.
[202] J. Antony, S. Pendyala, A. Sharma, X. B. Chen, J. Morrison, L.
Bergman, Y. Qiang, J. Appl. Phys. 2005, 97, 10D307.
[203] W. Lee, M. C. Jeong, S. W. Joo, J. M. Myoung, Nanotechnology
2005, 16, 764.
[204] A. Ishizumi, Y. Kanemitsu, Appl. Phys. Lett. 2005, 86, 253106.
[205] G. Z. Shen, J. H. Cho, J. K. Yoo, G. C. Yi, C. J. Lee, J. Phys. Chem.
B 2005, 109, 5491.
[206] H. Y. Xu, Y. C. Liu, R. Mu, C. L. Shao, Y. M. Lu, D. Z. Shen, X. W.
Fan, Appl. Phys. Lett. 2005, 86, 123107.
[207] S. M. Zhou, X. H. Zhang, X. M. Meng, S. K. Wu, S. T. Lee, Phys.
Stat. Sol. A 2005, 202, 405.
[208] X. S. Fang, C. H. Ye, L. D. Zhang, Y. Li, Z. D. Xiao, Chem. Lett.
2005, 34, 436.
[209] C. C. Lin, H. P. Chen, S. Y. Chen, Chem. Phys. Lett. 2005, 404,
30.
[210] S. Y. Bae, C. W. Na, J. H. Kang, J. Park, J. Phys. Chem. B 2005,
109, 2526.
[211] L. W. Yang, X. L. Wu, G. S. Huang, T. Qiu, Y. M. Yang, J. Appl.
Phys. 2005, 97, 014308.
[212] G. Z. Shen, J. H. Cho, S. I. Jung, C. J. Lee, Chem. Phys. Lett.
2005, 401, 529.
[213] S. M. Zhou, X. H. Zhang, X. M. Meng, X. Fan, S. K. Wu, S. T. Lee,
Physica E 2005, 25, 587.
[214] W. Lee, M. C. Jeong, J. M. Myoung, Appl. Phys. Lett. 2004, 85,
6167.
[215] H. Y. Wei, Y. S. Wu, L. L. Wu, C. X. Hu, Mater. Lett. 2005, 59,
271.
[216] J. S. Jie, G. Z. Wang, X. H. Han, J. G. Hou, J. Phys. Chem. B
2004, 108, 17027.
[217] B. C. Cheng, Y. H. Xiao, G. S. Wu, L. D. Zhang, Adv. Funct.
Mater. 2004, 14, 913.
[218] S. M. Zhou, X. H. Zhang, X. M. Meng, K. Zou, X. Fan, S. K. Wu,
S. T. Lee, Nanotechnology 2004, 15, 1152.
[219] S. Y. Li, P. Lin, C. Y. Lee, T. Y. Tseng, C. J. Huang, J. Phys. D 2004,
37, 2274.
[220] C. X. Xu, X. W. Sun, X. H. Zhang, L. Ke, S. J. Chua, Nanotechnol-
ogy 2004, 15, 856.
[221] S. Y. Bae, H. W. Seo, J. H. Park, J. Phys. Chem. B 2004, 108,
5206.
[222] J. S. Jie, G. Z. Wang, X. H. Han, Q. X. Yu, Y. Liao, G. P. Li, J. G.
Hou, Chem. Phys. Lett. 2004, 387, 466.
[223] V. A. L. Roy, A. B. Djurisic, H. Liu, X. X. Zhang, Y. H. Leung,
M. H. Xie, J. Gao, H. F. Lui, C. Surya, Appl. Phys. Lett. 2004, 84,
756.
[224] C. Ronning, P. X. Gao, Y. Ding, Z. L. Wang, D. Schwen, Appl.
Phys. Lett. 2004, 84, 783.
[225] T. Agne, Z. Guan, X. M. Li, H. Wolf, T. Wichert, H. Natter, R.
Hempelmann, Appl. Phys. Lett. 2003, 83, 1204.
[226] B. Y. Geng, G. Z. Wang, Z. Jiang, T. Xie, S. H. Sun, G. W. Meng,
L. D. Zhang, Appl. Phys. Lett. 2003, 82, 4791.
[227] X. T. Zhang, Y. C. Liu, J. Y. Zhang, Y. M. Lu, D. Z. Shen, X. W. Fan,
X. G. Kong, J. Cryst. Growth 2003, 254, 80.
[228] S. M. Liu, F. Q. Liu, Z. G. Wang, Chem. Phys. Lett. 2001, 343,
489.
[229] S. M. Liu, F. Q. Liu, H. Q. Guo, Z. H. Zhang, Z. G. Wang, Phys.
Lett. A 2000, 271, 128.
Received: March 16, 2006
Published online on July 18, 2006
small 2006, 2, No. 8-9, 944 961 2006 Wiley-VCH Verlag GmbH&Co. KGaA, Weinheim www.small-journal.com 961
Optical Properties of ZnO Nanostructures
You might also like
- Materials Characterisation ExercisesDocument4 pagesMaterials Characterisation Exercisesyaswanth1992No ratings yet
- SEM N TEM in Polymer CharacterizationDocument25 pagesSEM N TEM in Polymer CharacterizationNabihah AbdullahNo ratings yet
- X-Ray Fluorescence: Analytical BackgroundDocument18 pagesX-Ray Fluorescence: Analytical BackgroundJoy RoyNo ratings yet
- Slide Uv VisDocument54 pagesSlide Uv VisElka Sushea IINo ratings yet
- Fourier Transform Infrared SpectrosDocument27 pagesFourier Transform Infrared Spectrosrmarin_90No ratings yet
- X-Ray Photoelectron Spectroscopy (XPS) : Electron Spectroscopy For Chemical Analysis (ESCA)Document24 pagesX-Ray Photoelectron Spectroscopy (XPS) : Electron Spectroscopy For Chemical Analysis (ESCA)Imran KhanNo ratings yet
- Nano LithographyDocument14 pagesNano LithographyMohammad RameezNo ratings yet
- Carbon Nanotubes: Centre For Nanoscience and Technology, Pondicherry University Puducherry-605 014, IndiaDocument62 pagesCarbon Nanotubes: Centre For Nanoscience and Technology, Pondicherry University Puducherry-605 014, IndiaMohammad RameezNo ratings yet
- Source: KDI Co .LTD, 2011Document5 pagesSource: KDI Co .LTD, 2011Siti NurshahiraNo ratings yet
- T.Josphin Anto Monisha Ii-Msc - ChemistryDocument15 pagesT.Josphin Anto Monisha Ii-Msc - ChemistryJoantoNo ratings yet
- UV-Vis Spectroscopy: Chm622-Advance Organic SpectrosDocument50 pagesUV-Vis Spectroscopy: Chm622-Advance Organic Spectrossharifah sakinah syed soffianNo ratings yet
- FTIR Spectros PDFDocument3 pagesFTIR Spectros PDFRidaSirtaDewiTRNo ratings yet
- Optical Properties of MaterialsDocument18 pagesOptical Properties of MaterialsAshish Manatosh BarikNo ratings yet
- Various Tools and Techniques Used For Structural ElucidationDocument28 pagesVarious Tools and Techniques Used For Structural ElucidationAvinashNo ratings yet
- 2.1 XpsDocument59 pages2.1 XpsMaheep upadhyayNo ratings yet
- AFM PresentationDocument16 pagesAFM PresentationSulficker AliNo ratings yet
- Lecture 12 - Fiber Optic Communication - Semiconductor Laser PDFDocument46 pagesLecture 12 - Fiber Optic Communication - Semiconductor Laser PDFBruno Garcia TejadaNo ratings yet
- Antiferromagnetic MaterialsDocument3 pagesAntiferromagnetic MaterialssamarthNo ratings yet
- Electron Microscopy of Materials ReviewDocument27 pagesElectron Microscopy of Materials ReviewMaria Paula Fernandez JimenezNo ratings yet
- Ir SpectrometersDocument10 pagesIr SpectrometersVinícius FavrettoNo ratings yet
- Electron MicrosDocument11 pagesElectron MicrosAbir RoyNo ratings yet
- HolographyDocument17 pagesHolographymanoj kumar rout100% (2)
- UNIT IV NMR Mass - WatermarkDocument23 pagesUNIT IV NMR Mass - WatermarkvickyNo ratings yet
- X-Ray DiffractionDocument30 pagesX-Ray DiffractionMerve Ayvaz KöroğluNo ratings yet
- Atomic StructureDocument49 pagesAtomic StructureFatimaNo ratings yet
- Ruby Laser and CO2 Laser - PowerPointToPdfDocument15 pagesRuby Laser and CO2 Laser - PowerPointToPdfchirag pandeyNo ratings yet
- VoltammetryDocument13 pagesVoltammetryNandhanNo ratings yet
- Essay RamanDocument3 pagesEssay RamanChu Wai Seng100% (1)
- Dye Sensitized Solar Cells PresentationDocument20 pagesDye Sensitized Solar Cells PresentationAnand NaikNo ratings yet
- Ellipsometry: Alborg NiversityDocument138 pagesEllipsometry: Alborg Niversityanju guptaNo ratings yet
- Tissue Optics Light Scattering Methods and InstrumDocument7 pagesTissue Optics Light Scattering Methods and InstrumDaniela UrreaNo ratings yet
- 2 SynthesisDocument33 pages2 SynthesisSukanya Choudhary100% (1)
- Spectral Lines: Existence of Discrete Energy Levels Emission and Absorption SpectraDocument69 pagesSpectral Lines: Existence of Discrete Energy Levels Emission and Absorption SpectraACSVNo ratings yet
- Lecture 3 CVDDocument43 pagesLecture 3 CVDSam StideNo ratings yet
- Photoacoustic SpectrosDocument50 pagesPhotoacoustic Spectrosid.danlard5282No ratings yet
- Fluorescence MicrosDocument33 pagesFluorescence MicrosfatemaNo ratings yet
- Seminar On Photoconductive MaterialsDocument18 pagesSeminar On Photoconductive Materialssubhra3s50% (2)
- SaaDocument41 pagesSaaAbdur RahmanNo ratings yet
- Nanosheet 09179023Document7 pagesNanosheet 09179023ck maitiNo ratings yet
- Solid State Lasers and Applns - RKDocument56 pagesSolid State Lasers and Applns - RKAbhishek KumbalurNo ratings yet
- SuperconductivityDocument48 pagesSuperconductivityGiuseppe AromatarisNo ratings yet
- Optical Properties & CharacteristicsDocument21 pagesOptical Properties & CharacteristicsRogelyn JosolNo ratings yet
- Properties of Nano MaterialsDocument27 pagesProperties of Nano Materialsosamahiep100% (2)
- Optical Fibers: Structures, Optical Fibers: Structures, Waveguiding & FabricationDocument99 pagesOptical Fibers: Structures, Optical Fibers: Structures, Waveguiding & FabricationNung NingNo ratings yet
- 10 X-Ray DiffractionDocument8 pages10 X-Ray DiffractionProf.Dr.Mohamed Fahmy Mohamed Hussein100% (1)
- Photon Interaction With MatterDocument16 pagesPhoton Interaction With MatterShafuan WanNo ratings yet
- Voltammetric Techniques by Samuel P. KounavesDocument18 pagesVoltammetric Techniques by Samuel P. KounavesHiTuXNo ratings yet
- Short Overview of Perovskite Solar CellsDocument14 pagesShort Overview of Perovskite Solar CellsFAISAL TAIMURINo ratings yet
- PlasmaTech 3 TypesDocument34 pagesPlasmaTech 3 TypeswahidqhosyimNo ratings yet
- Optical MicrosDocument15 pagesOptical MicrosDHASARAIAH SNEHA100% (1)
- 3 SB LawDocument2 pages3 SB LawSukhwinder Singh GillNo ratings yet
- Flame PhotometerDocument18 pagesFlame PhotometerKuzhandai VeluNo ratings yet
- Chapter 3c X Ray DiffractionDocument48 pagesChapter 3c X Ray DiffractionmethoxyNo ratings yet
- Laser Types-Lecture (Dr. M Fadhali)Document29 pagesLaser Types-Lecture (Dr. M Fadhali)Mohamed Fadhali100% (1)
- Unit 4 Raman SpectrosDocument22 pagesUnit 4 Raman SpectrosNathanianNo ratings yet
- Alternative Experiment 4. Cyclic Voltammetry. IntroductionDocument7 pagesAlternative Experiment 4. Cyclic Voltammetry. IntroductionatulNo ratings yet
- Electroanalysis: Theory and Applications in Aqueous and Non-Aqueous Media and in Automated Chemical ControlFrom EverandElectroanalysis: Theory and Applications in Aqueous and Non-Aqueous Media and in Automated Chemical ControlNo ratings yet
- Thin Film Micro-Optics: New Frontiers of Spatio-Temporal Beam ShapingFrom EverandThin Film Micro-Optics: New Frontiers of Spatio-Temporal Beam ShapingNo ratings yet
- MIG Welding Class NotesDocument21 pagesMIG Welding Class NotesRaju SharmaNo ratings yet
- FSL106HRDocument13 pagesFSL106HRGerson SoaresNo ratings yet
- PWM Signal GenerationDocument4 pagesPWM Signal GenerationPravat SatpathyNo ratings yet
- Principles of Electronics RedoneDocument153 pagesPrinciples of Electronics RedoneOdale MitchellNo ratings yet
- Bab 7 - 1Document18 pagesBab 7 - 1Ivan FadillahNo ratings yet
- Full Chapter Nanomaterials Theory Problems and Solutions 2Nd Edition Upendranath Nandi Debnarayan Jana PDFDocument53 pagesFull Chapter Nanomaterials Theory Problems and Solutions 2Nd Edition Upendranath Nandi Debnarayan Jana PDFjohn.small350100% (3)
- Skin Response Meter: Sameer Salam B.E 3Rd Year, Ece-A Roll No:160309735050Document1 pageSkin Response Meter: Sameer Salam B.E 3Rd Year, Ece-A Roll No:160309735050Sameer SalamNo ratings yet
- Specification IC DK1203Document6 pagesSpecification IC DK1203EulerMartinsDeMello0% (1)
- Remote CodesDocument21 pagesRemote CodesChampika MadhurabhashinNo ratings yet
- Load CommutationDocument27 pagesLoad CommutationSobiNo ratings yet
- Design of Embedded ProcessorsDocument80 pagesDesign of Embedded ProcessorsSayan Kumar KhanNo ratings yet
- Tesis Finalcorrections PDFDocument166 pagesTesis Finalcorrections PDFdaniej25No ratings yet
- Wiring Diagrams For Rotary Phase ConvertorDocument2 pagesWiring Diagrams For Rotary Phase Convertorwhsprz100% (1)
- Rectifier Filter Spreadsheet Instructions PDFDocument7 pagesRectifier Filter Spreadsheet Instructions PDFUnderc OnstructionNo ratings yet
- Chemistry of Superconductor Materials Preparation, Chemistry, Characterization and Theory, Vanderah, 839PpDocument839 pagesChemistry of Superconductor Materials Preparation, Chemistry, Characterization and Theory, Vanderah, 839PpRay FraustoNo ratings yet
- Crystal OscillatorDocument3 pagesCrystal OscillatorsunilNo ratings yet
- High Voltage LinesDocument7 pagesHigh Voltage LinesvelisbarNo ratings yet
- Lecture - 1 Power AmplifiersDocument75 pagesLecture - 1 Power AmplifiersMadhumitha VetrivelNo ratings yet
- Physics ProjectDocument8 pagesPhysics ProjectXigfon SqitroNo ratings yet
- What Is DebouncingDocument7 pagesWhat Is DebouncingDimuthu Nuwan AbeysingheNo ratings yet
- Datasheet 24c01 EepromDocument26 pagesDatasheet 24c01 EepromItalo Vasquez LeonNo ratings yet
- Photonic CrystalsDocument53 pagesPhotonic CrystalsRafi AlifNo ratings yet
- Cost AnalysisDocument9 pagesCost AnalysisOkiPetrus Hutauruk LumbanBaringinNo ratings yet
- 140g br001 - en PDocument12 pages140g br001 - en PTanveer AhmedNo ratings yet
- Ac DC HarmonicDocument6 pagesAc DC Harmonicajeetdhakar3065No ratings yet
- ModelDocument2 pagesModelAnbalagan GuruNo ratings yet
- Eee 3204Document22 pagesEee 3204Sarwar Hosen SimonNo ratings yet
- RF MixersDocument29 pagesRF MixersName100% (10)
- Investigation Effect of Aluminum Atom On The Structural, Electronic and Optical Properties of Graphene Nanomaterials Using DFTDocument5 pagesInvestigation Effect of Aluminum Atom On The Structural, Electronic and Optical Properties of Graphene Nanomaterials Using DFTInternational Journal of Innovative Science and Research TechnologyNo ratings yet