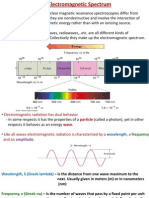
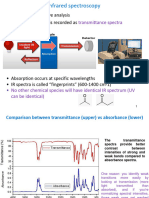
Download as pdf or txt
You might also like
- JBL Audio Engineering for Sound ReinforcementFrom EverandJBL Audio Engineering for Sound ReinforcementRating: 5 out of 5 stars5/5 (2)
- 4.11 Structure DeterminationDocument46 pages4.11 Structure DeterminationJaslinder Kaur Dhillon100% (1)
- Infrared SpectrosDocument9 pagesInfrared Spectrosbac_nobita7657No ratings yet
- Infrared SpectrosDocument4 pagesInfrared Spectrosjellybean07100% (1)
- Session 7 (IR)Document27 pagesSession 7 (IR)ashenafiNo ratings yet
- Ftir SpectrosDocument36 pagesFtir SpectrosHafiz Mudaser AhmadNo ratings yet
- CHEM 200 IR SpectrosDocument35 pagesCHEM 200 IR SpectrosIngrid LacsonNo ratings yet
- Infra RedDocument7 pagesInfra RedDennianasNo ratings yet
- Characteristic IR Absorption Frequencies of Organic Functional GroupsDocument19 pagesCharacteristic IR Absorption Frequencies of Organic Functional GroupsChandra Reddy100% (1)
- Infrared Spectroscopy: Conformational IsomersDocument7 pagesInfrared Spectroscopy: Conformational IsomersRiyan NazarudinNo ratings yet
- Infrared SpectrosDocument17 pagesInfrared SpectrosJasper DumalaogNo ratings yet
- Textile PhysicsDocument11 pagesTextile PhysicsAl AminNo ratings yet
- Infrared SpectrosDocument14 pagesInfrared SpectrosV G Viju KumarNo ratings yet
- FullDocument10 pagesFullAbdul Wahab KhanNo ratings yet
- Raw Material Analysis-IRDocument58 pagesRaw Material Analysis-IRDilla Wulan NingrumNo ratings yet
- Option Dancing WritingDocument17 pagesOption Dancing Writingsourabh kalkarNo ratings yet
- IR SpectrosDocument39 pagesIR SpectrosYurîiGislãineNo ratings yet
- INTRODUCTION To IR 2Document21 pagesINTRODUCTION To IR 2Rabia KhanNo ratings yet
- IR SPECTROSCOPY Notes FullDocument5 pagesIR SPECTROSCOPY Notes FullKartik KuteNo ratings yet
- Caligan Probset1Document5 pagesCaligan Probset1Jeff Tristan CaliganNo ratings yet
- SpectrosDocument39 pagesSpectrosJames Baben0% (1)
- Infrared SpectrosDocument9 pagesInfrared SpectrosMakeedaNo ratings yet
- Chap 12ADocument72 pagesChap 12Arareair213No ratings yet
- IR Spectroscopy IntroductionDocument14 pagesIR Spectroscopy IntroductionKasani Tirumala tejaNo ratings yet
- Organic Compound Identification Using Infrared SpectrosDocument34 pagesOrganic Compound Identification Using Infrared SpectrosRohit SinghNo ratings yet
- 1CHAPTER 1 Spectroscopy of C CompoundDocument47 pages1CHAPTER 1 Spectroscopy of C CompoundEustance JuanNo ratings yet
- Infraredspectroscopy 161018121240Document33 pagesInfraredspectroscopy 161018121240Rajkishor YadavNo ratings yet
- Infra Red SpectroscopiDocument42 pagesInfra Red SpectroscopiMade PrimaNo ratings yet
- IR SpectraDocument5 pagesIR SpectraXyphrus EmeritusNo ratings yet
- ASS Instrumental OrganicDocument17 pagesASS Instrumental OrganicMohamed SakrNo ratings yet
- Infrared SpectroscopyDocument12 pagesInfrared Spectroscopysanjay sNo ratings yet
- Spectroscopy IR NMR Supplemental ReadingDocument14 pagesSpectroscopy IR NMR Supplemental Readingmayagal1707No ratings yet
- Kuliah Infra Merah-LengkapDocument63 pagesKuliah Infra Merah-LengkapM Nur M. MahmudNo ratings yet
- Chap 12 - IRDocument30 pagesChap 12 - IRTanvir AhmedNo ratings yet
- Chem 212: Physical Analytical ChemistryDocument22 pagesChem 212: Physical Analytical ChemistrysamuelNo ratings yet
- Lecture 4 IR Spectrum AnalysisDocument43 pagesLecture 4 IR Spectrum AnalysiskhadijahhannahNo ratings yet
- Organic 7Document41 pagesOrganic 7sm737744No ratings yet
- Infrared Spectroscopy or Vibrational SpectrosDocument18 pagesInfrared Spectroscopy or Vibrational SpectrosVignesh Raja PNo ratings yet
- IR SpectrosDocument17 pagesIR SpectrosQasim Jalali NanotiNo ratings yet
- Spectroscopy - Chemistry.nmr - FTIR.ms. .SilversteinDocument476 pagesSpectroscopy - Chemistry.nmr - FTIR.ms. .SilversteinOscar PamosNo ratings yet
- Infrared Spectroscopy EceDocument50 pagesInfrared Spectroscopy EceRavi KumarNo ratings yet
- Infrared SpectrosDocument10 pagesInfrared SpectrosVal's Very OwnNo ratings yet
- IR OrganometallicDocument21 pagesIR OrganometallicYanti Yana HalidNo ratings yet
- Unit IV IR SpectroscoptyDocument37 pagesUnit IV IR SpectroscoptyNTGNo ratings yet
- Chapter 4 - FtirDocument45 pagesChapter 4 - Ftirmimi azmnNo ratings yet
- Introduction Interpretation of Spectra andDocument29 pagesIntroduction Interpretation of Spectra andDr. M. AslamNo ratings yet
- Notes 14C IRDocument6 pagesNotes 14C IRLouie SyNo ratings yet
- IR Lecture UpdateDocument141 pagesIR Lecture UpdateAnonymous epaORjlkgNo ratings yet
- Lecture 3.1 - Introduction To The Synthesis of Nanomaterials - Molecular SpectrosDocument88 pagesLecture 3.1 - Introduction To The Synthesis of Nanomaterials - Molecular SpectrosGian BanaresNo ratings yet
- An Introduction To Infrared and UV-Visible SpectrosDocument45 pagesAn Introduction To Infrared and UV-Visible SpectrosArvandz_tea100% (1)
- IR and NMR SpectrosDocument13 pagesIR and NMR SpectrosAnand BarapatreNo ratings yet
- Elusidasi Struktur Senyawa Organik-Minggu 2 Dan 3-FTIRDocument56 pagesElusidasi Struktur Senyawa Organik-Minggu 2 Dan 3-FTIRmuhammademiralyavich9876No ratings yet
- Wike Kusuma Wardani A. Title of Experiment: Infrared Spectroscopy B. The Aim of Experiment: Identify The Functional Groups Contained in ADocument10 pagesWike Kusuma Wardani A. Title of Experiment: Infrared Spectroscopy B. The Aim of Experiment: Identify The Functional Groups Contained in AWikeKusumaNo ratings yet
- Spectroscopy IRDocument33 pagesSpectroscopy IRNoor FatimaNo ratings yet
- UV-Visible and Ir SpectrosDocument29 pagesUV-Visible and Ir Spectrosppj25945No ratings yet
- B.Tech Spectroscopy Module 5.4: Applications of IR SpectrosDocument16 pagesB.Tech Spectroscopy Module 5.4: Applications of IR SpectrosKAMALANo ratings yet
- Spectroscopy Ir NMR and UvDocument26 pagesSpectroscopy Ir NMR and UvHaider JalalNo ratings yet
- PHR410 Chapter 2Document36 pagesPHR410 Chapter 2pulock.paulNo ratings yet
- Encyclopaedia Britannica, 11th Edition, Volume 6, Slice 8 "Conduction, Electric"From EverandEncyclopaedia Britannica, 11th Edition, Volume 6, Slice 8 "Conduction, Electric"No ratings yet
- Defects Activated Photoluminescence in Two-Dimensional Semiconductors: Interplay Between Bound, Charged, and Free ExcitonsDocument5 pagesDefects Activated Photoluminescence in Two-Dimensional Semiconductors: Interplay Between Bound, Charged, and Free ExcitonsHanh DuongNo ratings yet
- Al Khayat2016Document9 pagesAl Khayat2016Hanh DuongNo ratings yet
- Modern Organic Synthesis An IntroductionDocument37 pagesModern Organic Synthesis An IntroductionHanh DuongNo ratings yet
- Endeavour Postgraduate ScholarshipDocument1 pageEndeavour Postgraduate ScholarshipHanh DuongNo ratings yet
- Synthesis of PN-1Document1 pageSynthesis of PN-1Hanh DuongNo ratings yet
- TetrazoleSubstitutedPolymer Myhanh Modified 1Document17 pagesTetrazoleSubstitutedPolymer Myhanh Modified 1Hanh DuongNo ratings yet
- Students Understand The Basic Knowledge of Homogeneous, Heterogeneous, and PhotocatalystsDocument12 pagesStudents Understand The Basic Knowledge of Homogeneous, Heterogeneous, and PhotocatalystsHanh DuongNo ratings yet
- Something To ShareDocument10 pagesSomething To ShareHanh DuongNo ratings yet
- Intermediate PhasesDocument10 pagesIntermediate PhasesTommy HarzaNo ratings yet
- Lab Assignment 2Document7 pagesLab Assignment 2lasse.stiefelNo ratings yet
- Rajaratnam 199012345Document5 pagesRajaratnam 199012345Asim MazumderNo ratings yet
- All India Integrated Test Series: JEE (Advanced) - 2022Document11 pagesAll India Integrated Test Series: JEE (Advanced) - 2022Beyond ur imaginationNo ratings yet
- SIF3011 Tutorial2Document2 pagesSIF3011 Tutorial2Izzah RosliNo ratings yet
- Chapter 1Document1 pageChapter 1Naveed ZafarNo ratings yet
- Centrifugal ForceDocument11 pagesCentrifugal Forceno oneNo ratings yet
- Basic of FBC BoilerDocument25 pagesBasic of FBC BoilerJayam KondanNo ratings yet
- Epdm Rubber - Grade Yg-310Document1 pageEpdm Rubber - Grade Yg-310danielsu87No ratings yet
- GEOGRAPHY P1 GR10 QP NOV2018 - EnglishDocument16 pagesGEOGRAPHY P1 GR10 QP NOV2018 - EnglishAndzaniNo ratings yet
- Flashcards - Topic 6 Electrochemistry - CIE Chemistry A-LevelDocument83 pagesFlashcards - Topic 6 Electrochemistry - CIE Chemistry A-LevelSarah DevilNo ratings yet
- Mahoney Table SampleDocument11 pagesMahoney Table SampleVinoth KumarNo ratings yet
- Plaubel Makina 67 ReviewDocument5 pagesPlaubel Makina 67 ReviewkamonexNo ratings yet
- CDS VAM® FJL 11.75in. 60lb-ft P110 Alt. Drift 10.625in. 87.5%Document1 pageCDS VAM® FJL 11.75in. 60lb-ft P110 Alt. Drift 10.625in. 87.5%secanggang.scgdNo ratings yet
- CHM207 Lab Report Eks.3Document8 pagesCHM207 Lab Report Eks.3Akmal HakimNo ratings yet
- Metallurgy of Carbon SteelDocument5 pagesMetallurgy of Carbon SteelMadhavan SoundararajanNo ratings yet
- Q-Deformed Lie Algebras and Fractional CalculusDocument8 pagesQ-Deformed Lie Algebras and Fractional CalculusFahd ElshemaryNo ratings yet
- Heat Balance SheetDocument5 pagesHeat Balance SheetNiteesh Dua0% (1)
- Homework+4 20220511Document2 pagesHomework+4 20220511許廷睿No ratings yet
- Modulation Solutions For Nematicon PropagationDocument10 pagesModulation Solutions For Nematicon PropagationIsmael ArceNo ratings yet
- Elektronik Spektrum Dari Molekul DiatomikDocument6 pagesElektronik Spektrum Dari Molekul DiatomikNiesy0% (1)
- 04 Meteorological Applications SMDocument12 pages04 Meteorological Applications SMNavinNo ratings yet
- Reading 3 Week 16Document19 pagesReading 3 Week 16otgoo 0624No ratings yet
- 07 Calde Cast MW 150Document3 pages07 Calde Cast MW 150Abdul SabirNo ratings yet
- Turbomachines Unit IV PPT (Axial Flow Compressors & Steam Turbines)Document93 pagesTurbomachines Unit IV PPT (Axial Flow Compressors & Steam Turbines)SujanNo ratings yet
- Discussion, Explaintatition Dan ReviewDocument19 pagesDiscussion, Explaintatition Dan ReviewFadlan BarcelonistanNo ratings yet
- Lecture1 Part2 ME4227Document46 pagesLecture1 Part2 ME4227Thant Zaw AungNo ratings yet
- Tarea de SinusoidalesDocument2 pagesTarea de SinusoidalesRodrigo Nicolas VALENCIA VARGASNo ratings yet
- Studies of Welded Joints: Archives of Foundry EngineeringDocument6 pagesStudies of Welded Joints: Archives of Foundry EngineeringnkchandruNo ratings yet
- ADJECTIVEDocument13 pagesADJECTIVEEddowardho Otniel LegesangNo ratings yet