
Venu July07
Venu July07
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5822)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- Tim Horton CaseDocument12 pagesTim Horton CaseRafael Guerra OcampoNo ratings yet
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Terex RT 780. 80 Ton RTDocument20 pagesTerex RT 780. 80 Ton RTBhavana Kewlani67% (3)
- Lydia Amir - Plato's Theory of LoveDocument9 pagesLydia Amir - Plato's Theory of Lovediegochillo5506No ratings yet
- SPR S12 F SolDocument5 pagesSPR S12 F SolCengiz KayaNo ratings yet
- Sensors: License Plate Recognition Algorithm For Passenger Cars in Chinese Residential AreasDocument16 pagesSensors: License Plate Recognition Algorithm For Passenger Cars in Chinese Residential AreasCengiz KayaNo ratings yet
- ELEC6111: Detection and Estimation Theory Minimax Hypothesis TestingDocument17 pagesELEC6111: Detection and Estimation Theory Minimax Hypothesis TestingCengiz KayaNo ratings yet
- Course Objective: ELEC6111: Detection and Estimation TheoryDocument28 pagesCourse Objective: ELEC6111: Detection and Estimation TheoryCengiz KayaNo ratings yet
- Synchronous Buck Multiphase Optimized LGA Power Block: Integrated Power Semiconductors, Drivers & PassivesDocument10 pagesSynchronous Buck Multiphase Optimized LGA Power Block: Integrated Power Semiconductors, Drivers & PassivesCengiz KayaNo ratings yet
- ALL-IDB - The Acute Lymphoblastic Leukemia Image Database For Image ProcessingDocument5 pagesALL-IDB - The Acute Lymphoblastic Leukemia Image Database For Image ProcessingCengiz Kaya100% (1)
- Material and DesignDocument2 pagesMaterial and DesignrafadannNo ratings yet
- Phytosome: Presented byDocument14 pagesPhytosome: Presented bySari RamadhaniNo ratings yet
- Alfonso Lopez CardiovascularDocument15 pagesAlfonso Lopez Cardiovascularjaniceli0207100% (1)
- REX012832GYAP3N0Document28 pagesREX012832GYAP3N0marius.chitigaNo ratings yet
- Physics: InstructionsDocument8 pagesPhysics: InstructionsGracious SakaNo ratings yet
- GEA Spray DryerDocument4 pagesGEA Spray DryerDego Yusa AliNo ratings yet
- CB Unit 2Document67 pagesCB Unit 2Hardik KhandharNo ratings yet
- Sacred Places of The World A Religious Journey Across The GlobeDocument209 pagesSacred Places of The World A Religious Journey Across The GlobeDenis-Elena Cotea100% (1)
- Integrated Pest Management For Corn (Zea MaysDocument21 pagesIntegrated Pest Management For Corn (Zea MayscholzNo ratings yet
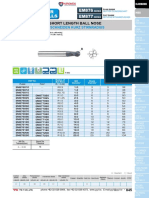
- X-Power End Mills EM877 EM876: Carbide, 2 Flute Short Length Ball NoseDocument2 pagesX-Power End Mills EM877 EM876: Carbide, 2 Flute Short Length Ball NosesahNo ratings yet
- Ayurvedic Herb - EKSHUDocument4 pagesAyurvedic Herb - EKSHUSanjay PisharodiNo ratings yet
- Feubo Katalog 2016Document44 pagesFeubo Katalog 2016Suelen BarbosaNo ratings yet
- Integrar 430 AutopilotDocument4 pagesIntegrar 430 Autopilotnelson vasquezNo ratings yet
- Factors Affecting ProductivityDocument16 pagesFactors Affecting ProductivityArslan MunawarNo ratings yet
- Alberts-Chapter 11-Membrane TransportDocument44 pagesAlberts-Chapter 11-Membrane TransportMayra SanchezNo ratings yet
- Gelman TaylorDocument443 pagesGelman TaylorAbhishek Bajaj100% (1)
- Mirroring Men and Masculinity in Joanne Harris' Chocolat and The Lollipop ShoesDocument50 pagesMirroring Men and Masculinity in Joanne Harris' Chocolat and The Lollipop ShoesBasneen Hudha AshrafNo ratings yet
- EME PPT Unit-5 - ElectiveDocument37 pagesEME PPT Unit-5 - ElectivesudeepkoreaNo ratings yet
- Organic Chemistry With Biological Applications 3Rd Edition Mcmurry Test Bank Full Chapter PDFDocument36 pagesOrganic Chemistry With Biological Applications 3Rd Edition Mcmurry Test Bank Full Chapter PDFmisstepmonocarp1b69100% (11)
- The Rotator Cuff (Myofascial Techniques)Document4 pagesThe Rotator Cuff (Myofascial Techniques)Advanced-Trainings.com100% (3)
- PLAINCRETE Blocks Matibay Pa!: Load Bearing Reinforced Concrete Blocks Money ConstructontheDocument7 pagesPLAINCRETE Blocks Matibay Pa!: Load Bearing Reinforced Concrete Blocks Money ConstructontheJim Bryan RazNo ratings yet
- Final Demo Lesson Plan Olermo Bonifacio Jr. C. 1Document17 pagesFinal Demo Lesson Plan Olermo Bonifacio Jr. C. 1juniobendiegoNo ratings yet
- Metabolisme Protein Dan Asam Amino: Mind Map TM Ii/Idk IDocument1 pageMetabolisme Protein Dan Asam Amino: Mind Map TM Ii/Idk ISania qurrataNo ratings yet
- Countable and Uncountable NounsDocument7 pagesCountable and Uncountable NounsValentine NNo ratings yet
- THE BANAO BODONG ASSOCIATION (BBA) SMALL SCALE GOLD MINING IN GA-ANG MINES TALALANG, BALBALAN KALINGA CAR, PHILIPPINES by ROYCE LINGBAWANDocument22 pagesTHE BANAO BODONG ASSOCIATION (BBA) SMALL SCALE GOLD MINING IN GA-ANG MINES TALALANG, BALBALAN KALINGA CAR, PHILIPPINES by ROYCE LINGBAWANpopsky100% (1)
- New Text DocumentDocument4 pagesNew Text DocumentGzim DragushaNo ratings yet
- Lab 1a - ME3000 Practical Op-Amp Circuits - v1.32Document7 pagesLab 1a - ME3000 Practical Op-Amp Circuits - v1.32MrmouzinhoNo ratings yet












































































Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5822)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- Tim Horton CaseDocument12 pagesTim Horton CaseRafael Guerra OcampoNo ratings yet
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Terex RT 780. 80 Ton RTDocument20 pagesTerex RT 780. 80 Ton RTBhavana Kewlani67% (3)
- Lydia Amir - Plato's Theory of LoveDocument9 pagesLydia Amir - Plato's Theory of Lovediegochillo5506No ratings yet
- SPR S12 F SolDocument5 pagesSPR S12 F SolCengiz KayaNo ratings yet
- Sensors: License Plate Recognition Algorithm For Passenger Cars in Chinese Residential AreasDocument16 pagesSensors: License Plate Recognition Algorithm For Passenger Cars in Chinese Residential AreasCengiz KayaNo ratings yet
- ELEC6111: Detection and Estimation Theory Minimax Hypothesis TestingDocument17 pagesELEC6111: Detection and Estimation Theory Minimax Hypothesis TestingCengiz KayaNo ratings yet
- Course Objective: ELEC6111: Detection and Estimation TheoryDocument28 pagesCourse Objective: ELEC6111: Detection and Estimation TheoryCengiz KayaNo ratings yet
- Synchronous Buck Multiphase Optimized LGA Power Block: Integrated Power Semiconductors, Drivers & PassivesDocument10 pagesSynchronous Buck Multiphase Optimized LGA Power Block: Integrated Power Semiconductors, Drivers & PassivesCengiz KayaNo ratings yet
- ALL-IDB - The Acute Lymphoblastic Leukemia Image Database For Image ProcessingDocument5 pagesALL-IDB - The Acute Lymphoblastic Leukemia Image Database For Image ProcessingCengiz Kaya100% (1)
- Material and DesignDocument2 pagesMaterial and DesignrafadannNo ratings yet
- Phytosome: Presented byDocument14 pagesPhytosome: Presented bySari RamadhaniNo ratings yet
- Alfonso Lopez CardiovascularDocument15 pagesAlfonso Lopez Cardiovascularjaniceli0207100% (1)
- REX012832GYAP3N0Document28 pagesREX012832GYAP3N0marius.chitigaNo ratings yet
- Physics: InstructionsDocument8 pagesPhysics: InstructionsGracious SakaNo ratings yet
- GEA Spray DryerDocument4 pagesGEA Spray DryerDego Yusa AliNo ratings yet
- CB Unit 2Document67 pagesCB Unit 2Hardik KhandharNo ratings yet
- Sacred Places of The World A Religious Journey Across The GlobeDocument209 pagesSacred Places of The World A Religious Journey Across The GlobeDenis-Elena Cotea100% (1)
- Integrated Pest Management For Corn (Zea MaysDocument21 pagesIntegrated Pest Management For Corn (Zea MayscholzNo ratings yet
- X-Power End Mills EM877 EM876: Carbide, 2 Flute Short Length Ball NoseDocument2 pagesX-Power End Mills EM877 EM876: Carbide, 2 Flute Short Length Ball NosesahNo ratings yet
- Ayurvedic Herb - EKSHUDocument4 pagesAyurvedic Herb - EKSHUSanjay PisharodiNo ratings yet
- Feubo Katalog 2016Document44 pagesFeubo Katalog 2016Suelen BarbosaNo ratings yet
- Integrar 430 AutopilotDocument4 pagesIntegrar 430 Autopilotnelson vasquezNo ratings yet
- Factors Affecting ProductivityDocument16 pagesFactors Affecting ProductivityArslan MunawarNo ratings yet
- Alberts-Chapter 11-Membrane TransportDocument44 pagesAlberts-Chapter 11-Membrane TransportMayra SanchezNo ratings yet
- Gelman TaylorDocument443 pagesGelman TaylorAbhishek Bajaj100% (1)
- Mirroring Men and Masculinity in Joanne Harris' Chocolat and The Lollipop ShoesDocument50 pagesMirroring Men and Masculinity in Joanne Harris' Chocolat and The Lollipop ShoesBasneen Hudha AshrafNo ratings yet
- EME PPT Unit-5 - ElectiveDocument37 pagesEME PPT Unit-5 - ElectivesudeepkoreaNo ratings yet
- Organic Chemistry With Biological Applications 3Rd Edition Mcmurry Test Bank Full Chapter PDFDocument36 pagesOrganic Chemistry With Biological Applications 3Rd Edition Mcmurry Test Bank Full Chapter PDFmisstepmonocarp1b69100% (11)
- The Rotator Cuff (Myofascial Techniques)Document4 pagesThe Rotator Cuff (Myofascial Techniques)Advanced-Trainings.com100% (3)
- PLAINCRETE Blocks Matibay Pa!: Load Bearing Reinforced Concrete Blocks Money ConstructontheDocument7 pagesPLAINCRETE Blocks Matibay Pa!: Load Bearing Reinforced Concrete Blocks Money ConstructontheJim Bryan RazNo ratings yet
- Final Demo Lesson Plan Olermo Bonifacio Jr. C. 1Document17 pagesFinal Demo Lesson Plan Olermo Bonifacio Jr. C. 1juniobendiegoNo ratings yet
- Metabolisme Protein Dan Asam Amino: Mind Map TM Ii/Idk IDocument1 pageMetabolisme Protein Dan Asam Amino: Mind Map TM Ii/Idk ISania qurrataNo ratings yet
- Countable and Uncountable NounsDocument7 pagesCountable and Uncountable NounsValentine NNo ratings yet
- THE BANAO BODONG ASSOCIATION (BBA) SMALL SCALE GOLD MINING IN GA-ANG MINES TALALANG, BALBALAN KALINGA CAR, PHILIPPINES by ROYCE LINGBAWANDocument22 pagesTHE BANAO BODONG ASSOCIATION (BBA) SMALL SCALE GOLD MINING IN GA-ANG MINES TALALANG, BALBALAN KALINGA CAR, PHILIPPINES by ROYCE LINGBAWANpopsky100% (1)
- New Text DocumentDocument4 pagesNew Text DocumentGzim DragushaNo ratings yet
- Lab 1a - ME3000 Practical Op-Amp Circuits - v1.32Document7 pagesLab 1a - ME3000 Practical Op-Amp Circuits - v1.32MrmouzinhoNo ratings yet