Download as pdf or txt
You might also like
- Unit 2 Progress Check: FRQDocument6 pagesUnit 2 Progress Check: FRQerwin golovashkinNo ratings yet
- X-Ray and VUV Spectra From The Laser Plasma Produced With "Kanal-2" FacilityDocument6 pagesX-Ray and VUV Spectra From The Laser Plasma Produced With "Kanal-2" FacilitytsrifNo ratings yet
- Rural News, Mar 2011Document4 pagesRural News, Mar 2011emediageNo ratings yet
- Ebenezer 2002 Curr Science OctDocument8 pagesEbenezer 2002 Curr Science OctDuraisingh Diamond EbenezerNo ratings yet
- Journal of Electroanalytical Chemistry: Hatem M.A. Amin, Yuki Uchida, Enno Kätelhön, Richard G. ComptonDocument7 pagesJournal of Electroanalytical Chemistry: Hatem M.A. Amin, Yuki Uchida, Enno Kätelhön, Richard G. ComptonSiti AmirahNo ratings yet
- Longitudinal Profile Diagnostic Scheme With Subfemtosecond Resolution For High-Brightness Electron BeamsDocument8 pagesLongitudinal Profile Diagnostic Scheme With Subfemtosecond Resolution For High-Brightness Electron BeamsParticle Beam Physics LabNo ratings yet
- 1984 - MORSE - MICHALOPOULOS - Chem - Rev - Spectroscopic Studies of The Jet-Cooled Nickel DimerDocument7 pages1984 - MORSE - MICHALOPOULOS - Chem - Rev - Spectroscopic Studies of The Jet-Cooled Nickel DimerAlejandra AwimbaweNo ratings yet
- Basics of Dynamic ElectrochemistryDocument15 pagesBasics of Dynamic ElectrochemistryNaresh Chavan50% (2)
- Characterization of The Plasma Column Used in Studies of A Plasma-Loaded Relativistic Backward Wave OscillatorDocument1 pageCharacterization of The Plasma Column Used in Studies of A Plasma-Loaded Relativistic Backward Wave Oscillatorvashisht druvaNo ratings yet
- Lan 1999Document7 pagesLan 1999maytco84No ratings yet
- Math Modeling ACEODocument14 pagesMath Modeling ACEOwei weiNo ratings yet
- Optical Transition Radiation: 'Work Supported Physics DepartmentDocument35 pagesOptical Transition Radiation: 'Work Supported Physics DepartmentParticle Beam Physics LabNo ratings yet
- Riemann Zeta Function and Hydrogen SpectrumDocument24 pagesRiemann Zeta Function and Hydrogen SpectrumIVAN ILIEV100% (1)
- Paper 3Document7 pagesPaper 3naveenbabu19No ratings yet
- Significance of Small Voltage in ImpedanceDocument6 pagesSignificance of Small Voltage in ImpedanceSingu Sai Vamsi Teja ee23m059No ratings yet
- Plasma Dynamics in An Arc Formed PDFDocument22 pagesPlasma Dynamics in An Arc Formed PDFsujayan2005No ratings yet
- Mathematical Analysis of A Zn/Niooh CellDocument6 pagesMathematical Analysis of A Zn/Niooh CellupscNo ratings yet
- AC ResistivityDocument6 pagesAC ResistivityJulio TedescoNo ratings yet
- Theoretical and Experimental Analysis of Porous Electrodes For Lithium-Ion Batteries by Electrochemical Impedance Spectroscopy Using A Symmetric CellDocument7 pagesTheoretical and Experimental Analysis of Porous Electrodes For Lithium-Ion Batteries by Electrochemical Impedance Spectroscopy Using A Symmetric Cellsaismaran999No ratings yet
- Plasma Simulation by Artificial Dielectrics and Parallel-Plate Media-Xy9Document14 pagesPlasma Simulation by Artificial Dielectrics and Parallel-Plate Media-Xy9archie222222No ratings yet
- Y. Yamakita Et Al - Controlling The Motion of Rydberg Molecules in Inhomogeneous Electric FieldDocument3 pagesY. Yamakita Et Al - Controlling The Motion of Rydberg Molecules in Inhomogeneous Electric FieldItama23No ratings yet
- E E Q E Q F: Solution of Problem 1Document9 pagesE E Q E Q F: Solution of Problem 1nurullah_bulutNo ratings yet
- Zavitaev 2006Document7 pagesZavitaev 2006Mario RectoNo ratings yet
- Zitterbewegung ExperimentDocument5 pagesZitterbewegung ExperimentAdonai CruzNo ratings yet
- Numerical Computation External Resonant Cavities: E.g., Coaxial Lines and WaveguidesDocument4 pagesNumerical Computation External Resonant Cavities: E.g., Coaxial Lines and WaveguideshmalrizzoNo ratings yet
- Electrical Excitation of Graphene PlasmonsDocument22 pagesElectrical Excitation of Graphene PlasmonsKOJANo ratings yet
- ION Electron Re Combination in Moderate Pressure Neon Discharge 1973-2-VinogradovDocument4 pagesION Electron Re Combination in Moderate Pressure Neon Discharge 1973-2-VinogradovPeter-sagamiNo ratings yet
- Circular WaveguideDocument19 pagesCircular WaveguideLam DinhNo ratings yet
- First Principles Theory of The EPR G-Tensor in Solids: Defects in QuartzDocument5 pagesFirst Principles Theory of The EPR G-Tensor in Solids: Defects in Quartzfghsda123000No ratings yet
- Electrical Properties of The Membranes of The Pleuropneumonia-Like Organism A 5969Document13 pagesElectrical Properties of The Membranes of The Pleuropneumonia-Like Organism A 5969Anjana's WorldNo ratings yet
- Ijetr022484 PDFDocument4 pagesIjetr022484 PDFsonawaneulhas292No ratings yet
- Investigation of Diverse Characteristics of Strained III-V Nitride Quantum WellDocument4 pagesInvestigation of Diverse Characteristics of Strained III-V Nitride Quantum WellUlhasNo ratings yet
- Generalized Sagdeev Approach To Nonlinear Plasma ExcitationsDocument11 pagesGeneralized Sagdeev Approach To Nonlinear Plasma ExcitationsDiego VegaNo ratings yet
- Eikonal Waves, Caustics and Mode Conversion in Tokamak PlasmasDocument26 pagesEikonal Waves, Caustics and Mode Conversion in Tokamak PlasmasEstrella MedinaNo ratings yet
- Jamel - EUROCORR 2008Document10 pagesJamel - EUROCORR 2008Ricardo NogueiraNo ratings yet
- Fundamentals of Atomic and Molecular Spectroscopy in Instrumental AnalysisDocument40 pagesFundamentals of Atomic and Molecular Spectroscopy in Instrumental Analysisnikusrivastva44043No ratings yet
- Voltammetry at A Microdisk ElectrodeDocument16 pagesVoltammetry at A Microdisk ElectrodeFelipe Cepeda SilvaNo ratings yet
- Parametric Study of The CuBr Laser With Hydrogen AdditivesDocument9 pagesParametric Study of The CuBr Laser With Hydrogen AdditivesDimoNo ratings yet
- Energy States of MoleculesDocument12 pagesEnergy States of MoleculesBenjamín Marc Ridgway de SassouNo ratings yet
- Electrochemical Impedance SpectrosDocument25 pagesElectrochemical Impedance SpectrosBogdan Buu100% (1)
- Cole HydrogenDocument15 pagesCole HydrogenDyra KesumaNo ratings yet
- Selfoscilstion Avslanche Article - 82Document3 pagesSelfoscilstion Avslanche Article - 82Mono De Tres CabezasNo ratings yet
- Characterization of A Spheromak Plasma Gun: The Effect of Refractory Electrode CoatingsDocument11 pagesCharacterization of A Spheromak Plasma Gun: The Effect of Refractory Electrode CoatingsFederico GaleanoNo ratings yet
- Electron Paramagnetic ResonanceDocument47 pagesElectron Paramagnetic Resonance150819850No ratings yet
- Actoydrazsing Electron Spin Resonance SpectrometerDocument4 pagesActoydrazsing Electron Spin Resonance SpectrometerSanketDesaiNo ratings yet
- Sensors: Plasmonic Nanostructures For Nano-Scale Bio-SensingDocument23 pagesSensors: Plasmonic Nanostructures For Nano-Scale Bio-SensingMahima TyagiNo ratings yet
- Redalyc: Sistema de Información CientíficaDocument9 pagesRedalyc: Sistema de Información CientíficaJ Jesús Villanueva GarcíaNo ratings yet
- Cense NE 215 Session1 Aug2014Document37 pagesCense NE 215 Session1 Aug2014Tara VishinNo ratings yet
- Expo InstruDocument5 pagesExpo InstruKevin Anthony Oré MaldonadoNo ratings yet
- Determination Effective Material Parameters For A Metamaterial Based Analysis of Local FieldsDocument4 pagesDetermination Effective Material Parameters For A Metamaterial Based Analysis of Local FieldsNageswara Rao ChallaNo ratings yet
- Theory of Square Wave TryDocument4 pagesTheory of Square Wave TryChandrashekar Vishwanath VishwanathNo ratings yet
- Complex Permittivity Measurement of Substrates Using Ring ResonatorDocument6 pagesComplex Permittivity Measurement of Substrates Using Ring ResonatorShridhar MathadNo ratings yet
- Simple Opamp-Based Circuit For Measuring Electro-Conductivity of Electrolytic Solutions in Hydroponics SystemDocument6 pagesSimple Opamp-Based Circuit For Measuring Electro-Conductivity of Electrolytic Solutions in Hydroponics SystemMohammadjawad BaratiNo ratings yet
- M. Pelc Et Al - Massless Fermions Generated by Ultrashort Laser PulsesDocument6 pagesM. Pelc Et Al - Massless Fermions Generated by Ultrashort Laser PulsesPocxaNo ratings yet
- N.E. Andreev Et Al - Beat-Wave Experiments in The Micro Wave Range: Pump DepletionDocument4 pagesN.E. Andreev Et Al - Beat-Wave Experiments in The Micro Wave Range: Pump DepletionVasmazxNo ratings yet
- Determination of The Band Gap of A Semiconductor by Four Probe Set-UpDocument10 pagesDetermination of The Band Gap of A Semiconductor by Four Probe Set-UpMohit SharmaNo ratings yet
- E R Williams - New Experimental Test of Coulomb-S LawDocument40 pagesE R Williams - New Experimental Test of Coulomb-S LawNestor GasparNo ratings yet
- Jandieris - Temporal Power Spectrum of Scattered Electromagnetic Waves in The Equatorial Terrestrial IonosphereDocument12 pagesJandieris - Temporal Power Spectrum of Scattered Electromagnetic Waves in The Equatorial Terrestrial IonosphereOleg KharshiladzeNo ratings yet
- Edward L Hamilton, Chris H Greene and H R Sadeghpour - Shape-Resonance-Induced Long-Range Molecular Rydberg StatesDocument8 pagesEdward L Hamilton, Chris H Greene and H R Sadeghpour - Shape-Resonance-Induced Long-Range Molecular Rydberg StatesItama23No ratings yet
- Feynman Lectures Simplified 2C: Electromagnetism: in Relativity & in Dense MatterFrom EverandFeynman Lectures Simplified 2C: Electromagnetism: in Relativity & in Dense MatterNo ratings yet
- Verifying A Virtual Component Interface-Based PCI Bus Wrapper Using An LSC-Based SpecificationDocument59 pagesVerifying A Virtual Component Interface-Based PCI Bus Wrapper Using An LSC-Based SpecificationMehanathan Maggie MikeyNo ratings yet
- National Institute of Technology, Hamirpur 177 005 (HP) : Application Form For Faculty Position On Contract BasisDocument2 pagesNational Institute of Technology, Hamirpur 177 005 (HP) : Application Form For Faculty Position On Contract BasisMehanathan Maggie MikeyNo ratings yet
- 1856.typical Signal Chain Solutions For IndustrialDocument64 pages1856.typical Signal Chain Solutions For IndustrialMehanathan Maggie MikeyNo ratings yet
- An Ultra-Low Quiescent Current CMOS Low-Dropout Regulator With Small Output Voltage VariationsDocument6 pagesAn Ultra-Low Quiescent Current CMOS Low-Dropout Regulator With Small Output Voltage VariationsMehanathan Maggie MikeyNo ratings yet
- VLSI Design ManualDocument96 pagesVLSI Design ManualMehanathan Maggie MikeyNo ratings yet
- Fsd0a A Generic Core Prodbrief v1.0Document1 pageFsd0a A Generic Core Prodbrief v1.0Mehanathan Maggie MikeyNo ratings yet
- Phaselock Techniques (Gardner-2005)Document254 pagesPhaselock Techniques (Gardner-2005)Mehanathan Maggie Mikey100% (1)
- Railway Security Through The Use of Wireless Sensor Networks Based On Fuzzy LogicDocument11 pagesRailway Security Through The Use of Wireless Sensor Networks Based On Fuzzy LogicMehanathan Maggie MikeyNo ratings yet
- 0804Document41 pages0804Mehanathan Maggie MikeyNo ratings yet
- Image Processing Using FPGADocument12 pagesImage Processing Using FPGAMehanathan Maggie MikeyNo ratings yet
- Chapitre 1 1Document15 pagesChapitre 1 1Ri HabNo ratings yet
- Topic 10 Organic Chemistry 10.1 To 10.2 20.1 To 20.3Document120 pagesTopic 10 Organic Chemistry 10.1 To 10.2 20.1 To 20.3Supriyaa ChordiaNo ratings yet
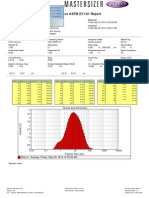
- Result: Sieve ASTM E11:61 Report: 1.837 D (0.9) : 34.676 D (0.5) : D (0.1) : 8.637 Um Um UmDocument1 pageResult: Sieve ASTM E11:61 Report: 1.837 D (0.9) : 34.676 D (0.5) : D (0.1) : 8.637 Um Um UmAmina MujkanovicNo ratings yet
- Structure of Atom Class 11Document1 pageStructure of Atom Class 11VikNo ratings yet
- AEE 463 Homework 12Document5 pagesAEE 463 Homework 12Andy TranNo ratings yet
- EXPT 3 - DryingDocument10 pagesEXPT 3 - DryingCharlyn Joy RamirezNo ratings yet
- Precipitation Titrimetry-221Document11 pagesPrecipitation Titrimetry-221HudzaifiNo ratings yet
- A Review of Solid Electrolyte Interphase SEI and Dendrite Formation in Lithium Batteries 2023 SpringerDocument46 pagesA Review of Solid Electrolyte Interphase SEI and Dendrite Formation in Lithium Batteries 2023 SpringerCIKARANG KOTANo ratings yet
- Brochure Home Care Dissolvine m40 Ms Global enDocument20 pagesBrochure Home Care Dissolvine m40 Ms Global entony tangNo ratings yet
- Chemical KineticsDocument22 pagesChemical KineticsDarnlNo ratings yet
- Henley E.J., Seader J.D. - Equilibrium-Stage SeparationDocument384 pagesHenley E.J., Seader J.D. - Equilibrium-Stage SeparationYuri Kaminski83% (6)
- 5 Metamorphic RocksDocument20 pages5 Metamorphic RocksYounas BilalNo ratings yet
- Analysis of An Antacid Lab ReportDocument5 pagesAnalysis of An Antacid Lab ReportClandy CoNo ratings yet
- Science Class X Sample Paper Test 02 For Board Exam 2024 AnswersDocument14 pagesScience Class X Sample Paper Test 02 For Board Exam 2024 Answerssingh2008adityaNo ratings yet
- Corrosion and Corrosion ControlDocument43 pagesCorrosion and Corrosion ControlTEZ ANALYSIS AND STORIESNo ratings yet
- QLMN ND CourseCatalogueDocument2 pagesQLMN ND CourseCatalogueIsmail CherkaouiNo ratings yet
- M103-Conducting - Quantum TheroyDocument13 pagesM103-Conducting - Quantum Theroymades samiNo ratings yet
- Mtu Engine Model 20V4000G63L A. LT CIRCUIT (410 KW@ 30 M /HR) Heat Exchanger: Gl-13Lx36 Duty RequirementsDocument4 pagesMtu Engine Model 20V4000G63L A. LT CIRCUIT (410 KW@ 30 M /HR) Heat Exchanger: Gl-13Lx36 Duty RequirementsbaljeetjatNo ratings yet
- A Simulation Study of Heat Transfer in Polymerization ReactorsDocument15 pagesA Simulation Study of Heat Transfer in Polymerization ReactorsArcangelo Di TanoNo ratings yet
- 9701 s09 QP 2Document25 pages9701 s09 QP 2Hubbak KhanNo ratings yet
- Plate Heat ExchangerDocument1 pagePlate Heat ExchangerBvitalize100% (1)
- Solu BilityDocument23 pagesSolu BilityVidya PasalkarNo ratings yet
- G02 CM133L Experiment6Document6 pagesG02 CM133L Experiment6Alexandria Nicole CaalimNo ratings yet
- 11 Refrigeration CyclesDocument18 pages11 Refrigeration CyclesHussamNo ratings yet
- Plate - 3 (FLOT)Document2 pagesPlate - 3 (FLOT)patrick dgNo ratings yet
- Chemical Energetics 2 MSDocument4 pagesChemical Energetics 2 MSAli SiddiqNo ratings yet
- Chromatography: Mikhail TsvetDocument115 pagesChromatography: Mikhail TsvetAjinkya PuranikNo ratings yet
- Galvanic/Voltaic CellsDocument12 pagesGalvanic/Voltaic CellsEve BonellNo ratings yet
- LESSON 5 Chemical ReactionsDocument52 pagesLESSON 5 Chemical ReactionsFil HynneyNo ratings yet