
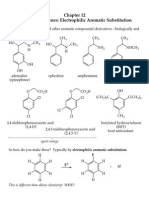
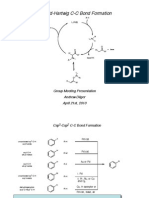
Chapter04 Oxidationd
Chapter04 Oxidationd
You might also like
- Lab Manual - Professor Dr. Stefan Surzycki (Auth.) - Basic Techniques in Molecular Biology-Springer-Verlag Berlin Heidelberg (2000) PDFDocument441 pagesLab Manual - Professor Dr. Stefan Surzycki (Auth.) - Basic Techniques in Molecular Biology-Springer-Verlag Berlin Heidelberg (2000) PDFCris Noroña100% (1)
- Asme NM.3.1-18 PDFDocument470 pagesAsme NM.3.1-18 PDFPablo DM100% (2)
- Schaum's Easy Outline of Organic Chemistry, Second EditionFrom EverandSchaum's Easy Outline of Organic Chemistry, Second EditionRating: 3.5 out of 5 stars3.5/5 (2)
- Asymmetric SynthesisDocument55 pagesAsymmetric Synthesisevsgoud_goud0% (1)
- Chemistry - Harvard's Advanced Organic Chemistry 2003Document717 pagesChemistry - Harvard's Advanced Organic Chemistry 2003ramik100% (23)
- Coursesaver (Chad) College Physics OutlinesDocument40 pagesCoursesaver (Chad) College Physics Outlinescalong558No ratings yet
- Nylon Polymers Book-Degradation and Stabilization of Nylon PolymersDocument120 pagesNylon Polymers Book-Degradation and Stabilization of Nylon PolymersWilliam H. BasingerNo ratings yet
- Fire Safety NotesDocument13 pagesFire Safety Notesrescueone93% (14)
- 05 Conformational Anal 2Document11 pages05 Conformational Anal 2Swati GautamNo ratings yet
- Chemistry 22: Some Notes On Chapter 10 Reactions: Y Y Y YDocument6 pagesChemistry 22: Some Notes On Chapter 10 Reactions: Y Y Y Yhairey947594No ratings yet
- Bi FunctionalDocument36 pagesBi FunctionalchidambaramrNo ratings yet
- Chen Feb 07Document9 pagesChen Feb 07Debdeep RayNo ratings yet
- 16 Cycloaddition Rxns 1Document13 pages16 Cycloaddition Rxns 1Aulia RhamdaniNo ratings yet
- Ferreira Lit 7-17-03Document14 pagesFerreira Lit 7-17-03bocahupiNo ratings yet
- Benzene and Aromatic Compounds-fazli-In ClassDocument57 pagesBenzene and Aromatic Compounds-fazli-In Classjokowi123No ratings yet
- Enola TesDocument21 pagesEnola TesReksy WibowoNo ratings yet
- Chapter 12Document23 pagesChapter 12Hải NguyễnNo ratings yet
- Answer Key: Chemistry 206 First Hour ExaminationDocument9 pagesAnswer Key: Chemistry 206 First Hour Examinationsudipta88No ratings yet
- Epoxidation & Opening PDFDocument84 pagesEpoxidation & Opening PDFRajkumar ChinnuNo ratings yet
- AKD BuchwaldhartwigDocument39 pagesAKD BuchwaldhartwigMurali Venkat NagNo ratings yet
- Hyper ConjugationDocument29 pagesHyper ConjugationDargorlethNo ratings yet
- Chemistry 206 Advanced Organic Chemistry: Olefin Addition Reactions: Part-2Document17 pagesChemistry 206 Advanced Organic Chemistry: Olefin Addition Reactions: Part-2eraborNo ratings yet
- Olefin Metathesis in Organic SynthesisDocument19 pagesOlefin Metathesis in Organic SynthesisaegosmithNo ratings yet
- CH 8 Handouts (All)Document34 pagesCH 8 Handouts (All)Ryan MaNo ratings yet
- Chemistry 206 Advanced Organic Chemistry: Chem 206 D. A. EvansDocument14 pagesChemistry 206 Advanced Organic Chemistry: Chem 206 D. A. EvanseraborNo ratings yet
- Aromatic ChemistryDocument16 pagesAromatic ChemistrysimbaleoNo ratings yet
- Problems On Named ReactionsDocument103 pagesProblems On Named ReactionsBapu ThoratNo ratings yet
- CH 8 Answers (All)Document33 pagesCH 8 Answers (All)Cham Nguyen0% (1)
- Chapter 12Document69 pagesChapter 12Pace AjjaNo ratings yet
- CHM 1321 Assignment 4 AnswersDocument12 pagesCHM 1321 Assignment 4 AnswersSara YuenNo ratings yet
- Photochemistry PDFDocument36 pagesPhotochemistry PDFAna RisticNo ratings yet
- AromaticDocument38 pagesAromaticDerrick Maatla MoadiNo ratings yet
- Neighbouring Group ParticipationDocument15 pagesNeighbouring Group ParticipationAbdulMananNo ratings yet
- Wilkinson CatalystDocument19 pagesWilkinson Catalystjagabandhu_patraNo ratings yet
- Muscone PDFDocument27 pagesMuscone PDFa d e eNo ratings yet
- Summary Sheet: Two Key Concepts For Nucleophilic Substitution On CarbonylsDocument1 pageSummary Sheet: Two Key Concepts For Nucleophilic Substitution On CarbonylsChitKoNo ratings yet
- Chem 215 Myers: Sharpless Asymmetric Dihydroxylation ReactionDocument4 pagesChem 215 Myers: Sharpless Asymmetric Dihydroxylation ReactionPrabhakar S AchantaNo ratings yet
- Alkenes and Alkynes II:: Addition ReactionsDocument102 pagesAlkenes and Alkynes II:: Addition Reactionsfingil20032003No ratings yet
- Chem 115 Myers: Shi Asymmetric Epoxidation ReactionDocument5 pagesChem 115 Myers: Shi Asymmetric Epoxidation ReactionashisdasNo ratings yet
- Organic ChemistryDocument41 pagesOrganic ChemistrySunil ChoudharyNo ratings yet
- Asymmetric Synthesis: α-Substitution using chiral enolatesDocument36 pagesAsymmetric Synthesis: α-Substitution using chiral enolatesrajjkfghNo ratings yet
- Final Exam KeyDocument12 pagesFinal Exam KeykitthiNo ratings yet
- Stereo SpecificDocument15 pagesStereo SpecificroxtrNo ratings yet
- CH 07Document96 pagesCH 07Jacquot AbendrothNo ratings yet
- CH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHDocument8 pagesCH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHShahbaz NazirNo ratings yet
- Lecture 1Document11 pagesLecture 1Fang GaoNo ratings yet
- Aromatic Compounds: Y Y Y YDocument9 pagesAromatic Compounds: Y Y Y YCamille AdleNo ratings yet
- 06 Conformational Anal 3Document13 pages06 Conformational Anal 3eraborNo ratings yet
- Effect of Structure On Reactivity of CompoundDocument8 pagesEffect of Structure On Reactivity of Compoundeagl33ye50% (2)
- CHM 1321 Assignment #2 - : AnswersDocument11 pagesCHM 1321 Assignment #2 - : AnswersSara YuenNo ratings yet
- Suzuky ReactionDocument13 pagesSuzuky ReactionAlbornoz JuanNo ratings yet
- Homogeneous Catalysis PDFDocument99 pagesHomogeneous Catalysis PDFevsgoud_goudNo ratings yet
- CH 23 StudentDocument33 pagesCH 23 StudentRabin ShresthaNo ratings yet
- Organic Chemistry Reactions BookDocument56 pagesOrganic Chemistry Reactions BookKeith Philippe100% (10)
- Answers To Chapter 1 In-Chapter ProblemsDocument13 pagesAnswers To Chapter 1 In-Chapter ProblemsChinmay DabkeNo ratings yet
- Exam 26030 F17Document8 pagesExam 26030 F17Christian CederhornNo ratings yet
- Summary Sheet 2: Enols and Enolates: Does Not FormDocument1 pageSummary Sheet 2: Enols and Enolates: Does Not FormAleestraNo ratings yet
- 23 Sigma Tropic Seminario Org IIIDocument19 pages23 Sigma Tropic Seminario Org IIIvaguinhoaquinoNo ratings yet
- Properties of SiliconDocument16 pagesProperties of SiliconGabriela ResendeNo ratings yet
- Retro Synthetic Analysis GuidelinesDocument12 pagesRetro Synthetic Analysis GuidelinesaukidoNo ratings yet
- The Total Synthesis of Natural ProductsFrom EverandThe Total Synthesis of Natural ProductsJohn ApSimonNo ratings yet
- Union v. Rick ScottDocument8 pagesUnion v. Rick ScottWilliam H. BasingerNo ratings yet
- Case: 10-11979 Date Filed: 06/16/2011 Page: 1 of 17: LLLLLLLLLLLLLLLLLLLLLDocument17 pagesCase: 10-11979 Date Filed: 06/16/2011 Page: 1 of 17: LLLLLLLLLLLLLLLLLLLLLWilliam H. BasingerNo ratings yet
- ROBERTSON v. HECKSEL Liberty Interest in Adult Son 11th-Circuit1260527Document6 pagesROBERTSON v. HECKSEL Liberty Interest in Adult Son 11th-Circuit1260527William H. BasingerNo ratings yet
- GEORGIA v. RANDOLPH 4th Amendment Violations - Roomate Allowed Search and Siezure Against The Accused Objections2Document51 pagesGEORGIA v. RANDOLPH 4th Amendment Violations - Roomate Allowed Search and Siezure Against The Accused Objections2William H. BasingerNo ratings yet
- President Bush ReportDocument2 pagesPresident Bush ReportWilliam H. BasingerNo ratings yet
- ContemptPacket Cobb CountyDocument11 pagesContemptPacket Cobb CountyWilliam H. BasingerNo ratings yet
- Tetraiodoethylene Sales Spec - AjayDocument1 pageTetraiodoethylene Sales Spec - AjayWilliam H. BasingerNo ratings yet
- Msds Aluminum StearateDocument4 pagesMsds Aluminum StearateWilliam H. BasingerNo ratings yet
- 2-Iodotoluene Spec 070903Document1 page2-Iodotoluene Spec 070903William H. BasingerNo ratings yet
- Uv FinishDocument67 pagesUv FinishChandra SekarNo ratings yet
- Fundamentals of Fluid MechanicsDocument1 pageFundamentals of Fluid MechanicsJuan Pablo Giraldo FrancoNo ratings yet
- A Brief History of CatalysisDocument9 pagesA Brief History of CatalysisKike BayonaNo ratings yet
- Next-Generation ABACUS Biosensors Reveal Cellular ABA Dynamics Driving Root Growth at Low Aerial HumidityDocument26 pagesNext-Generation ABACUS Biosensors Reveal Cellular ABA Dynamics Driving Root Growth at Low Aerial HumidityHùng HoàngNo ratings yet
- As 3870-1991 Certified Reference Materials - General Guide To Material Selection Preparation Testing and CertDocument7 pagesAs 3870-1991 Certified Reference Materials - General Guide To Material Selection Preparation Testing and CertSAI Global - APACNo ratings yet
- Ammonia TechnologyDocument13 pagesAmmonia TechnologyMihaela Popescu-NeagoeNo ratings yet
- 1A-Acrolon 890Document2 pages1A-Acrolon 890Stuart PhamNo ratings yet
- Concentration TechniquesDocument14 pagesConcentration TechniquesJoshua Rei NacpilNo ratings yet
- Science Form 4 Chapter 5 5.3Document44 pagesScience Form 4 Chapter 5 5.3KSSM TSENo ratings yet
- A Method To Test The Detectability of GC PFPD ForDocument8 pagesA Method To Test The Detectability of GC PFPD ForMarian Si Teofana HasnaNo ratings yet
- KULIAH BIOKIMIA DASAR - Dr. Chusnul HanimDocument51 pagesKULIAH BIOKIMIA DASAR - Dr. Chusnul HanimMuhammad JuhanNo ratings yet
- EnzymesDocument13 pagesEnzymesAna Liza DolomandingNo ratings yet
- Milroyal D With DSD Technology BrochureDocument12 pagesMilroyal D With DSD Technology Brochuredevil3300No ratings yet
- A Brief History of ThermodynamicsDocument17 pagesA Brief History of ThermodynamicsŞükrü_talaşNo ratings yet
- Real Gases - AnnotatedDocument43 pagesReal Gases - Annotatedrahulinder1234No ratings yet
- Bunker OilDocument5 pagesBunker OilJaymart Hernandez MojicaNo ratings yet
- Sterile Formulation MCQsDocument5 pagesSterile Formulation MCQsbalamurugan75% (8)
- Design Aspects of Tall Chimneys: S. N. ManoharDocument8 pagesDesign Aspects of Tall Chimneys: S. N. ManoharRama KrishnaNo ratings yet
- Shrinkage Cavity Analysis in Butterfly Valve Disc Castings IJERTV2IS111097Document8 pagesShrinkage Cavity Analysis in Butterfly Valve Disc Castings IJERTV2IS111097a.sutatNo ratings yet
- Powder Behaviour and The Nature of PowdersDocument6 pagesPowder Behaviour and The Nature of PowderspneuconNo ratings yet
- Line Sizing For Systems PDFDocument13 pagesLine Sizing For Systems PDFCua TranNo ratings yet
- The Effect of Bismuth On The Lead Acid Battery System (PDFDrive)Document255 pagesThe Effect of Bismuth On The Lead Acid Battery System (PDFDrive)Bores ModearNo ratings yet
- Org Synthesis QuizDocument71 pagesOrg Synthesis Quizlianchen251110100% (1)
- Solid Waste Incinerator ManufacturersDocument2 pagesSolid Waste Incinerator Manufacturersbl engineeringNo ratings yet
- Logistik Farmasi Saldo Awal Nama Bentuk/Satuan Hna+Ppn QTT JumlahDocument122 pagesLogistik Farmasi Saldo Awal Nama Bentuk/Satuan Hna+Ppn QTT JumlahListya HasanahNo ratings yet
- Ingersoll RandDocument10 pagesIngersoll RandJhonnyNo ratings yet
- Analisa Fizik SPMDocument1 pageAnalisa Fizik SPMkamalharmozaNo ratings yet


































































































Download as pdf or txt
You might also like
- Lab Manual - Professor Dr. Stefan Surzycki (Auth.) - Basic Techniques in Molecular Biology-Springer-Verlag Berlin Heidelberg (2000) PDFDocument441 pagesLab Manual - Professor Dr. Stefan Surzycki (Auth.) - Basic Techniques in Molecular Biology-Springer-Verlag Berlin Heidelberg (2000) PDFCris Noroña100% (1)
- Asme NM.3.1-18 PDFDocument470 pagesAsme NM.3.1-18 PDFPablo DM100% (2)
- Schaum's Easy Outline of Organic Chemistry, Second EditionFrom EverandSchaum's Easy Outline of Organic Chemistry, Second EditionRating: 3.5 out of 5 stars3.5/5 (2)
- Asymmetric SynthesisDocument55 pagesAsymmetric Synthesisevsgoud_goud0% (1)
- Chemistry - Harvard's Advanced Organic Chemistry 2003Document717 pagesChemistry - Harvard's Advanced Organic Chemistry 2003ramik100% (23)
- Coursesaver (Chad) College Physics OutlinesDocument40 pagesCoursesaver (Chad) College Physics Outlinescalong558No ratings yet
- Nylon Polymers Book-Degradation and Stabilization of Nylon PolymersDocument120 pagesNylon Polymers Book-Degradation and Stabilization of Nylon PolymersWilliam H. BasingerNo ratings yet
- Fire Safety NotesDocument13 pagesFire Safety Notesrescueone93% (14)
- 05 Conformational Anal 2Document11 pages05 Conformational Anal 2Swati GautamNo ratings yet
- Chemistry 22: Some Notes On Chapter 10 Reactions: Y Y Y YDocument6 pagesChemistry 22: Some Notes On Chapter 10 Reactions: Y Y Y Yhairey947594No ratings yet
- Bi FunctionalDocument36 pagesBi FunctionalchidambaramrNo ratings yet
- Chen Feb 07Document9 pagesChen Feb 07Debdeep RayNo ratings yet
- 16 Cycloaddition Rxns 1Document13 pages16 Cycloaddition Rxns 1Aulia RhamdaniNo ratings yet
- Ferreira Lit 7-17-03Document14 pagesFerreira Lit 7-17-03bocahupiNo ratings yet
- Benzene and Aromatic Compounds-fazli-In ClassDocument57 pagesBenzene and Aromatic Compounds-fazli-In Classjokowi123No ratings yet
- Enola TesDocument21 pagesEnola TesReksy WibowoNo ratings yet
- Chapter 12Document23 pagesChapter 12Hải NguyễnNo ratings yet
- Answer Key: Chemistry 206 First Hour ExaminationDocument9 pagesAnswer Key: Chemistry 206 First Hour Examinationsudipta88No ratings yet
- Epoxidation & Opening PDFDocument84 pagesEpoxidation & Opening PDFRajkumar ChinnuNo ratings yet
- AKD BuchwaldhartwigDocument39 pagesAKD BuchwaldhartwigMurali Venkat NagNo ratings yet
- Hyper ConjugationDocument29 pagesHyper ConjugationDargorlethNo ratings yet
- Chemistry 206 Advanced Organic Chemistry: Olefin Addition Reactions: Part-2Document17 pagesChemistry 206 Advanced Organic Chemistry: Olefin Addition Reactions: Part-2eraborNo ratings yet
- Olefin Metathesis in Organic SynthesisDocument19 pagesOlefin Metathesis in Organic SynthesisaegosmithNo ratings yet
- CH 8 Handouts (All)Document34 pagesCH 8 Handouts (All)Ryan MaNo ratings yet
- Chemistry 206 Advanced Organic Chemistry: Chem 206 D. A. EvansDocument14 pagesChemistry 206 Advanced Organic Chemistry: Chem 206 D. A. EvanseraborNo ratings yet
- Aromatic ChemistryDocument16 pagesAromatic ChemistrysimbaleoNo ratings yet
- Problems On Named ReactionsDocument103 pagesProblems On Named ReactionsBapu ThoratNo ratings yet
- CH 8 Answers (All)Document33 pagesCH 8 Answers (All)Cham Nguyen0% (1)
- Chapter 12Document69 pagesChapter 12Pace AjjaNo ratings yet
- CHM 1321 Assignment 4 AnswersDocument12 pagesCHM 1321 Assignment 4 AnswersSara YuenNo ratings yet
- Photochemistry PDFDocument36 pagesPhotochemistry PDFAna RisticNo ratings yet
- AromaticDocument38 pagesAromaticDerrick Maatla MoadiNo ratings yet
- Neighbouring Group ParticipationDocument15 pagesNeighbouring Group ParticipationAbdulMananNo ratings yet
- Wilkinson CatalystDocument19 pagesWilkinson Catalystjagabandhu_patraNo ratings yet
- Muscone PDFDocument27 pagesMuscone PDFa d e eNo ratings yet
- Summary Sheet: Two Key Concepts For Nucleophilic Substitution On CarbonylsDocument1 pageSummary Sheet: Two Key Concepts For Nucleophilic Substitution On CarbonylsChitKoNo ratings yet
- Chem 215 Myers: Sharpless Asymmetric Dihydroxylation ReactionDocument4 pagesChem 215 Myers: Sharpless Asymmetric Dihydroxylation ReactionPrabhakar S AchantaNo ratings yet
- Alkenes and Alkynes II:: Addition ReactionsDocument102 pagesAlkenes and Alkynes II:: Addition Reactionsfingil20032003No ratings yet
- Chem 115 Myers: Shi Asymmetric Epoxidation ReactionDocument5 pagesChem 115 Myers: Shi Asymmetric Epoxidation ReactionashisdasNo ratings yet
- Organic ChemistryDocument41 pagesOrganic ChemistrySunil ChoudharyNo ratings yet
- Asymmetric Synthesis: α-Substitution using chiral enolatesDocument36 pagesAsymmetric Synthesis: α-Substitution using chiral enolatesrajjkfghNo ratings yet
- Final Exam KeyDocument12 pagesFinal Exam KeykitthiNo ratings yet
- Stereo SpecificDocument15 pagesStereo SpecificroxtrNo ratings yet
- CH 07Document96 pagesCH 07Jacquot AbendrothNo ratings yet
- CH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHDocument8 pagesCH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHShahbaz NazirNo ratings yet
- Lecture 1Document11 pagesLecture 1Fang GaoNo ratings yet
- Aromatic Compounds: Y Y Y YDocument9 pagesAromatic Compounds: Y Y Y YCamille AdleNo ratings yet
- 06 Conformational Anal 3Document13 pages06 Conformational Anal 3eraborNo ratings yet
- Effect of Structure On Reactivity of CompoundDocument8 pagesEffect of Structure On Reactivity of Compoundeagl33ye50% (2)
- CHM 1321 Assignment #2 - : AnswersDocument11 pagesCHM 1321 Assignment #2 - : AnswersSara YuenNo ratings yet
- Suzuky ReactionDocument13 pagesSuzuky ReactionAlbornoz JuanNo ratings yet
- Homogeneous Catalysis PDFDocument99 pagesHomogeneous Catalysis PDFevsgoud_goudNo ratings yet
- CH 23 StudentDocument33 pagesCH 23 StudentRabin ShresthaNo ratings yet
- Organic Chemistry Reactions BookDocument56 pagesOrganic Chemistry Reactions BookKeith Philippe100% (10)
- Answers To Chapter 1 In-Chapter ProblemsDocument13 pagesAnswers To Chapter 1 In-Chapter ProblemsChinmay DabkeNo ratings yet
- Exam 26030 F17Document8 pagesExam 26030 F17Christian CederhornNo ratings yet
- Summary Sheet 2: Enols and Enolates: Does Not FormDocument1 pageSummary Sheet 2: Enols and Enolates: Does Not FormAleestraNo ratings yet
- 23 Sigma Tropic Seminario Org IIIDocument19 pages23 Sigma Tropic Seminario Org IIIvaguinhoaquinoNo ratings yet
- Properties of SiliconDocument16 pagesProperties of SiliconGabriela ResendeNo ratings yet
- Retro Synthetic Analysis GuidelinesDocument12 pagesRetro Synthetic Analysis GuidelinesaukidoNo ratings yet
- The Total Synthesis of Natural ProductsFrom EverandThe Total Synthesis of Natural ProductsJohn ApSimonNo ratings yet
- Union v. Rick ScottDocument8 pagesUnion v. Rick ScottWilliam H. BasingerNo ratings yet
- Case: 10-11979 Date Filed: 06/16/2011 Page: 1 of 17: LLLLLLLLLLLLLLLLLLLLLDocument17 pagesCase: 10-11979 Date Filed: 06/16/2011 Page: 1 of 17: LLLLLLLLLLLLLLLLLLLLLWilliam H. BasingerNo ratings yet
- ROBERTSON v. HECKSEL Liberty Interest in Adult Son 11th-Circuit1260527Document6 pagesROBERTSON v. HECKSEL Liberty Interest in Adult Son 11th-Circuit1260527William H. BasingerNo ratings yet
- GEORGIA v. RANDOLPH 4th Amendment Violations - Roomate Allowed Search and Siezure Against The Accused Objections2Document51 pagesGEORGIA v. RANDOLPH 4th Amendment Violations - Roomate Allowed Search and Siezure Against The Accused Objections2William H. BasingerNo ratings yet
- President Bush ReportDocument2 pagesPresident Bush ReportWilliam H. BasingerNo ratings yet
- ContemptPacket Cobb CountyDocument11 pagesContemptPacket Cobb CountyWilliam H. BasingerNo ratings yet
- Tetraiodoethylene Sales Spec - AjayDocument1 pageTetraiodoethylene Sales Spec - AjayWilliam H. BasingerNo ratings yet
- Msds Aluminum StearateDocument4 pagesMsds Aluminum StearateWilliam H. BasingerNo ratings yet
- 2-Iodotoluene Spec 070903Document1 page2-Iodotoluene Spec 070903William H. BasingerNo ratings yet
- Uv FinishDocument67 pagesUv FinishChandra SekarNo ratings yet
- Fundamentals of Fluid MechanicsDocument1 pageFundamentals of Fluid MechanicsJuan Pablo Giraldo FrancoNo ratings yet
- A Brief History of CatalysisDocument9 pagesA Brief History of CatalysisKike BayonaNo ratings yet
- Next-Generation ABACUS Biosensors Reveal Cellular ABA Dynamics Driving Root Growth at Low Aerial HumidityDocument26 pagesNext-Generation ABACUS Biosensors Reveal Cellular ABA Dynamics Driving Root Growth at Low Aerial HumidityHùng HoàngNo ratings yet
- As 3870-1991 Certified Reference Materials - General Guide To Material Selection Preparation Testing and CertDocument7 pagesAs 3870-1991 Certified Reference Materials - General Guide To Material Selection Preparation Testing and CertSAI Global - APACNo ratings yet
- Ammonia TechnologyDocument13 pagesAmmonia TechnologyMihaela Popescu-NeagoeNo ratings yet
- 1A-Acrolon 890Document2 pages1A-Acrolon 890Stuart PhamNo ratings yet
- Concentration TechniquesDocument14 pagesConcentration TechniquesJoshua Rei NacpilNo ratings yet
- Science Form 4 Chapter 5 5.3Document44 pagesScience Form 4 Chapter 5 5.3KSSM TSENo ratings yet
- A Method To Test The Detectability of GC PFPD ForDocument8 pagesA Method To Test The Detectability of GC PFPD ForMarian Si Teofana HasnaNo ratings yet
- KULIAH BIOKIMIA DASAR - Dr. Chusnul HanimDocument51 pagesKULIAH BIOKIMIA DASAR - Dr. Chusnul HanimMuhammad JuhanNo ratings yet
- EnzymesDocument13 pagesEnzymesAna Liza DolomandingNo ratings yet
- Milroyal D With DSD Technology BrochureDocument12 pagesMilroyal D With DSD Technology Brochuredevil3300No ratings yet
- A Brief History of ThermodynamicsDocument17 pagesA Brief History of ThermodynamicsŞükrü_talaşNo ratings yet
- Real Gases - AnnotatedDocument43 pagesReal Gases - Annotatedrahulinder1234No ratings yet
- Bunker OilDocument5 pagesBunker OilJaymart Hernandez MojicaNo ratings yet
- Sterile Formulation MCQsDocument5 pagesSterile Formulation MCQsbalamurugan75% (8)
- Design Aspects of Tall Chimneys: S. N. ManoharDocument8 pagesDesign Aspects of Tall Chimneys: S. N. ManoharRama KrishnaNo ratings yet
- Shrinkage Cavity Analysis in Butterfly Valve Disc Castings IJERTV2IS111097Document8 pagesShrinkage Cavity Analysis in Butterfly Valve Disc Castings IJERTV2IS111097a.sutatNo ratings yet
- Powder Behaviour and The Nature of PowdersDocument6 pagesPowder Behaviour and The Nature of PowderspneuconNo ratings yet
- Line Sizing For Systems PDFDocument13 pagesLine Sizing For Systems PDFCua TranNo ratings yet
- The Effect of Bismuth On The Lead Acid Battery System (PDFDrive)Document255 pagesThe Effect of Bismuth On The Lead Acid Battery System (PDFDrive)Bores ModearNo ratings yet
- Org Synthesis QuizDocument71 pagesOrg Synthesis Quizlianchen251110100% (1)
- Solid Waste Incinerator ManufacturersDocument2 pagesSolid Waste Incinerator Manufacturersbl engineeringNo ratings yet
- Logistik Farmasi Saldo Awal Nama Bentuk/Satuan Hna+Ppn QTT JumlahDocument122 pagesLogistik Farmasi Saldo Awal Nama Bentuk/Satuan Hna+Ppn QTT JumlahListya HasanahNo ratings yet
- Ingersoll RandDocument10 pagesIngersoll RandJhonnyNo ratings yet
- Analisa Fizik SPMDocument1 pageAnalisa Fizik SPMkamalharmozaNo ratings yet