Download as pdf or txt
You might also like
- Determining Electrical Conductivity Using The Electromagnetic (Eddy-Current) MethodDocument5 pagesDetermining Electrical Conductivity Using The Electromagnetic (Eddy-Current) MethodpablojorgesilvaNo ratings yet
- Metallic Oxides by GoodenoughDocument255 pagesMetallic Oxides by Goodenoughmuk_hawkNo ratings yet
- Catalysts: Structure of Nanocrystalline, Partially Disordered Mos Derived From Hrtem-An Abundant Material For EDocument16 pagesCatalysts: Structure of Nanocrystalline, Partially Disordered Mos Derived From Hrtem-An Abundant Material For EDuc Anh NguyenNo ratings yet
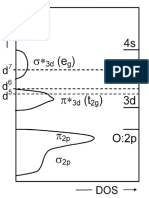
- Monoelectronic Band Structure For A Cubic PerovskiteDocument1 pageMonoelectronic Band Structure For A Cubic PerovskiteSJNo ratings yet
- Zwitterion ManuscriptDocument35 pagesZwitterion ManuscriptRishu KhuranaNo ratings yet
- Journal of American Ceramic SocietyDocument8 pagesJournal of American Ceramic SocietyNeeraj PanwarNo ratings yet
- Acetileno HidratasaDocument5 pagesAcetileno Hidratasacaanmaro17No ratings yet
- Asymmetry in Mössbauer Spectra of Fe - Azaporphyrin ComplexesDocument6 pagesAsymmetry in Mössbauer Spectra of Fe - Azaporphyrin ComplexesGudo SidhuNo ratings yet
- Accepted Manuscript: Inorganica Chimica ActaDocument13 pagesAccepted Manuscript: Inorganica Chimica Actamisranasrof9No ratings yet
- ESR and Magnetic Studies of OctahedralDocument8 pagesESR and Magnetic Studies of OctahedralYasmi Reyes OrtegaNo ratings yet
- Enhancement of Thermoelectric Properties of Porphyrin-Based 2021Document7 pagesEnhancement of Thermoelectric Properties of Porphyrin-Based 2021Toshar GargNo ratings yet
- Modulating Charge Patterning and Ionic Strength As A Strategy To Induce Conformational Changes in Intrinsically Disordered ProteinsDocument9 pagesModulating Charge Patterning and Ionic Strength As A Strategy To Induce Conformational Changes in Intrinsically Disordered ProteinsDCPNo ratings yet
- EPR M N - H I P: OF Ononuclear ON EME RON RoteinsDocument36 pagesEPR M N - H I P: OF Ononuclear ON EME RON RoteinsWilliam AgudeloNo ratings yet
- Crystals 11 01050 v2Document7 pagesCrystals 11 01050 v2Giovanni Samir Perez HerreraNo ratings yet
- Si:P As A Laboratory Analogue For Hydrogen On High Magnetic Field White Dwarf StarsDocument8 pagesSi:P As A Laboratory Analogue For Hydrogen On High Magnetic Field White Dwarf StarsAlex FlemingNo ratings yet
- Enhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryDocument3 pagesEnhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryJuaxmawNo ratings yet
- EsrDocument5 pagesEsrVirendra Singh Rajput100% (1)
- Acs Inorgchem 7b02272Document12 pagesAcs Inorgchem 7b02272SETIFI FatimaNo ratings yet
- Mishra Chem Mater 2010Document11 pagesMishra Chem Mater 2010MemoVillasenorNo ratings yet
- Ef 700500 XDocument6 pagesEf 700500 XMo OsNo ratings yet
- ESR For TY As HandoutDocument16 pagesESR For TY As HandoutPavitra JonesNo ratings yet
- Oxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3Document23 pagesOxygen Vacancies Effect On The Magnetic Properties of SrTiFeO3solisiusNo ratings yet
- 1 s2.0 S0277538721003818 MainDocument11 pages1 s2.0 S0277538721003818 MainMoromi NathNo ratings yet
- Effect of Yttrium Substitution On The Structural and Magnetic Properties of Smfeo3Document5 pagesEffect of Yttrium Substitution On The Structural and Magnetic Properties of Smfeo3rautsubhajit89No ratings yet
- Protein Binding and The Electronic Properties of Iron (II) Complexes: An Electrochemical and Optical Investigation of Outer Sphere EffectsDocument10 pagesProtein Binding and The Electronic Properties of Iron (II) Complexes: An Electrochemical and Optical Investigation of Outer Sphere EffectsDfmso0No ratings yet
- Origin of The Photoinduced Geometrical Change of Copper (I) Complexes From The Quantum Chemical Topology ViewDocument10 pagesOrigin of The Photoinduced Geometrical Change of Copper (I) Complexes From The Quantum Chemical Topology ViewJoakin BahamondesNo ratings yet
- Wyckoff PositionsDocument12 pagesWyckoff PositionsMatei100% (1)
- Transport Gap Renormalization at A Metal-Molecule Interface Using DFT-NEGF and Spin Unrestricted CalculationsDocument9 pagesTransport Gap Renormalization at A Metal-Molecule Interface Using DFT-NEGF and Spin Unrestricted CalculationsvijaykumarlambaNo ratings yet
- EddddDocument11 pagesEddddBRYAN ARIEL CRUZADO CARPIONo ratings yet
- Reactions of Laser-Ablated Zirconium Atoms Within A Supersonic Expansion: Insertion Versus Radical MechanismDocument11 pagesReactions of Laser-Ablated Zirconium Atoms Within A Supersonic Expansion: Insertion Versus Radical MechanismDiego Alejandro Hurtado BalcazarNo ratings yet
- RodioDocument5 pagesRodioTavo RodriguezNo ratings yet
- Density Functional Study of Gold and Iron Clusters On Perfect and Defected GrapheneDocument15 pagesDensity Functional Study of Gold and Iron Clusters On Perfect and Defected GrapheneTarzanNo ratings yet
- Solvent-Dependent Structure of Iridium Dihydride Complexes Different Geometries at Low and High Dielectricity of The MediumDocument10 pagesSolvent-Dependent Structure of Iridium Dihydride Complexes Different Geometries at Low and High Dielectricity of The Mediumxuyijing2007comNo ratings yet
- MC Cull Agh 2021Document11 pagesMC Cull Agh 2021muhammad ahmadNo ratings yet
- The Biology and Chemistry of High-Valent Iron-Oxo and Iron-Nitrido ComplexesDocument13 pagesThe Biology and Chemistry of High-Valent Iron-Oxo and Iron-Nitrido ComplexesMacky321No ratings yet
- Crystalline EnvironmentDocument11 pagesCrystalline Environmentdomehap737No ratings yet
- DenoDocument11 pagesDenoJames MartinNo ratings yet
- Real DFT Model On Co Adsorption by Noble CatalystDocument52 pagesReal DFT Model On Co Adsorption by Noble CatalystDikra BkNo ratings yet
- A Mathematical Model For The Porous Lead Dioxide ElectrodeDocument10 pagesA Mathematical Model For The Porous Lead Dioxide Electrodesumit singhNo ratings yet
- Prutt Ivar As in 2015Document10 pagesPrutt Ivar As in 2015Veerareddy VippalaNo ratings yet
- Photochemistry and Photophysics of Coordination Compounds of The Main Group MetalsDocument7 pagesPhotochemistry and Photophysics of Coordination Compounds of The Main Group MetalsгогавагановNo ratings yet
- Ncomms3817 PDFDocument6 pagesNcomms3817 PDFOussama IkhlefNo ratings yet
- Brodholt p1049-1053 97Document5 pagesBrodholt p1049-1053 97bencekeNo ratings yet
- Ncomms 13986Document6 pagesNcomms 13986Chiru MukherjeeNo ratings yet
- Macroscopic Transport by Synthetic Molecular Machines: ArticlesDocument7 pagesMacroscopic Transport by Synthetic Molecular Machines: ArticlesChristian j.No ratings yet
- Subject ChemistryDocument11 pagesSubject ChemistryErinle RahmatNo ratings yet
- R. Gonzalez-Ferez and P. Schmelcher - Rovibrational Spectra of Diatomic Molecules in Strong Electric Fields: The Adiabatic RegimeDocument11 pagesR. Gonzalez-Ferez and P. Schmelcher - Rovibrational Spectra of Diatomic Molecules in Strong Electric Fields: The Adiabatic RegimeImasmzNo ratings yet
- Direct Assignment of Molecular Vibrations Via Normal Mode Analysis of The Neutron Dynamic Pair Distribution Function TechniqueDocument24 pagesDirect Assignment of Molecular Vibrations Via Normal Mode Analysis of The Neutron Dynamic Pair Distribution Function TechniqueRabbia AminNo ratings yet
- Dense Plasma Temperature Equilibration in The Binary Collision ApproximationDocument5 pagesDense Plasma Temperature Equilibration in The Binary Collision ApproximationImperial Agent 5241No ratings yet
- 1999.06.15 - RUBAN - PRB - Surface Segregation TMalloysDocument12 pages1999.06.15 - RUBAN - PRB - Surface Segregation TMalloysAlejandra AwimbaweNo ratings yet
- Characterization of Acidity in ZSM-5 Zeolites: An X-Ray Photoelectron and I R Spectroscopy StudyDocument6 pagesCharacterization of Acidity in ZSM-5 Zeolites: An X-Ray Photoelectron and I R Spectroscopy StudyWulandariNo ratings yet
- Reactions of Alkanes and Reforming of NaphthaDocument95 pagesReactions of Alkanes and Reforming of NaphthaHenrique SouzaNo ratings yet
- Mechanistic Study of Methanol Synthesis From CO and H On A Modified Model Mo S ClusterDocument36 pagesMechanistic Study of Methanol Synthesis From CO and H On A Modified Model Mo S Clusterbalasekaran natarajanNo ratings yet
- Long Distance Electron Transfer in Cytochrome C Oxidase Immobilised On Electrodes. A Surface Enhanced Resonance Raman Spectroscopic StudyDocument8 pagesLong Distance Electron Transfer in Cytochrome C Oxidase Immobilised On Electrodes. A Surface Enhanced Resonance Raman Spectroscopic StudyEdward PittsNo ratings yet
- SSRN Id4367939Document29 pagesSSRN Id4367939ammar ghodbaniNo ratings yet
- Boosting The Performance of Single-Atom Catalysts Via External Electric Field PolarizationDocument12 pagesBoosting The Performance of Single-Atom Catalysts Via External Electric Field Polarizationnikusrivastva44043No ratings yet
- Study of The Underlying Electrochemistry of Polycrystalline GoldDocument13 pagesStudy of The Underlying Electrochemistry of Polycrystalline GoldAzucena osornio villaNo ratings yet
- Journal of Alloys and Compounds: M. Saidani, W. Belkacem, A. Bezergheanu, C.B. Cizmas, N. MlikiDocument10 pagesJournal of Alloys and Compounds: M. Saidani, W. Belkacem, A. Bezergheanu, C.B. Cizmas, N. MlikiHuckkey HuNo ratings yet
- 1 s2.0 S0927796X02001043 MainDocument56 pages1 s2.0 S0927796X02001043 MainpescaofritoNo ratings yet
- Crystal Field Theory (CFT)Document15 pagesCrystal Field Theory (CFT)veronicaNo ratings yet
- Progress in the Science and Technology of the Rare Earths: Volume 2From EverandProgress in the Science and Technology of the Rare Earths: Volume 2No ratings yet
- Ai-900 3df695e8afa1Document61 pagesAi-900 3df695e8afa1shaharpi24No ratings yet
- WebCruiser Web Vulnerability Scanner Test ReportDocument14 pagesWebCruiser Web Vulnerability Scanner Test ReportjhonassaNo ratings yet
- Audi Q7Document76 pagesAudi Q7spy2o67% (3)
- Chapter#03 Mcqs Project Management: PersonalityDocument11 pagesChapter#03 Mcqs Project Management: PersonalityIrfan GondalNo ratings yet
- Caking of Refined SugarDocument3 pagesCaking of Refined Sugarnghi100% (1)
- Python Self Study MaterialDocument9 pagesPython Self Study MaterialSrividhya ManikandanNo ratings yet
- Remote Control SQL DatabaseDocument25 pagesRemote Control SQL DatabaseSalwa QasemNo ratings yet
- Complience Sheet 10kvaDocument5 pagesComplience Sheet 10kvaNurye NigusNo ratings yet
- VectorDocument24 pagesVectorKshitij Bansal100% (1)
- Serial Schedule Non-Serial Schedule: CheckpointsDocument7 pagesSerial Schedule Non-Serial Schedule: CheckpointsSandeep BurmanNo ratings yet
- Lecture 1 Introduction and HistoryDocument58 pagesLecture 1 Introduction and HistoryDominiqueNo ratings yet
- Module 2 Fluiid StaticsDocument4 pagesModule 2 Fluiid StaticsVanvan BitonNo ratings yet
- Glass Standards (Draft) PDFDocument5 pagesGlass Standards (Draft) PDFCristian TofanNo ratings yet
- Mid-Infrared Spectroscopy As A Valuable Tool To TaDocument33 pagesMid-Infrared Spectroscopy As A Valuable Tool To TaThya efeNo ratings yet
- Week 6 Intro To Kernel Modules, Project 2: Sarah Diesburg Florida State UniversityDocument61 pagesWeek 6 Intro To Kernel Modules, Project 2: Sarah Diesburg Florida State Universityrobat2014No ratings yet
- UP - Organic ChemistryDocument14 pagesUP - Organic ChemistryKate EvangelistaNo ratings yet
- CISCO Built-In Wireshark Capability White Paper c11-554444Document10 pagesCISCO Built-In Wireshark Capability White Paper c11-554444Ramaswamy PeriaswamyNo ratings yet
- Antioxidant and Free Radical Scavenging Potential of Justicia Gendarussa Burm. Leaves in Vitro.Document8 pagesAntioxidant and Free Radical Scavenging Potential of Justicia Gendarussa Burm. Leaves in Vitro.Wrexford BritneyNo ratings yet
- Module 3: The BSI Symbols, Part I (C)Document5 pagesModule 3: The BSI Symbols, Part I (C)antcbeNo ratings yet
- How To Walk Bass Lines IV (#35) (Live Transcript)Document9 pagesHow To Walk Bass Lines IV (#35) (Live Transcript)Nicole CremeNo ratings yet
- Ecg303-M3-01 Soil Permeability and Seepage PDFDocument20 pagesEcg303-M3-01 Soil Permeability and Seepage PDFDk AshokNo ratings yet
- Pneumatic Water Pumping SystemDocument46 pagesPneumatic Water Pumping SystemAjithNo ratings yet
- LinuxCNC User ManualDocument208 pagesLinuxCNC User Manualgiovanne_torres5884No ratings yet
- Engineering ThermodynamicsDocument26 pagesEngineering ThermodynamicsRAJADURAI V (PA2211002010106)No ratings yet
- Carrier: Psychrometric ChartDocument1 pageCarrier: Psychrometric ChartAhmed SabryNo ratings yet
- Digital Keyboard: Service ManualDocument45 pagesDigital Keyboard: Service ManualsugedaNo ratings yet
- Chemistry Questions Mole ConceptDocument15 pagesChemistry Questions Mole ConceptbhaijanNo ratings yet
- Third Periodic Rating Math 10Document7 pagesThird Periodic Rating Math 10Sonny SamboNo ratings yet
- Productbrochure - L150G - L180G - L220G - VOE22A1006521 - 2011-02Document32 pagesProductbrochure - L150G - L180G - L220G - VOE22A1006521 - 2011-02Pradeep Raj100% (1)