Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5823)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Aerobic Digestion PDFDocument74 pagesAerobic Digestion PDFvalkiria112No ratings yet
- Electrochemical Study of Gold Cementation Onto CopperDocument2 pagesElectrochemical Study of Gold Cementation Onto CopperAFLAC ............100% (1)
- Cswip 1.0Document4 pagesCswip 1.0KianToro YeoNo ratings yet
- MC Injekt2033 PDFDocument2 pagesMC Injekt2033 PDFhemantrulzNo ratings yet
- Omegaven 1Document2 pagesOmegaven 1Devarajan Melpanaiyur SowmyanarayananNo ratings yet
- Report No: MSE/JEL/EXM/CRISP/PMI/F/001Document20 pagesReport No: MSE/JEL/EXM/CRISP/PMI/F/001Thiru RajaNo ratings yet
- Laboratory Errors in Analytical LaboratoryDocument30 pagesLaboratory Errors in Analytical LaboratorySathish Vemula100% (1)
- Loading & Discharge Hoses For Offshore MooringsDocument17 pagesLoading & Discharge Hoses For Offshore Mooringsศุภกฤต รักในหลวง100% (3)
- Environmental Pollution: Patricia L. GillisDocument7 pagesEnvironmental Pollution: Patricia L. GillisZemen TadegeNo ratings yet
- Journal of Industrial and Engineering ChemistryDocument6 pagesJournal of Industrial and Engineering ChemistryMarlen MayorgaNo ratings yet
- Estimation of Sugar in MilkDocument5 pagesEstimation of Sugar in MilkYunkai Day100% (1)
- Trymer ™ 2000 XP y FoamglassDocument3 pagesTrymer ™ 2000 XP y FoamglassCarlos Humberto Camarena CalvoNo ratings yet
- Huawei p-10 - GuidoDocument240 pagesHuawei p-10 - GuidoCristiNo ratings yet
- Product Safety Commission (Afps) : Management: Federal Institute For Occupational Safety and HealthDocument12 pagesProduct Safety Commission (Afps) : Management: Federal Institute For Occupational Safety and HealthFabricio AmorimNo ratings yet
- Kishimoto2021 2022Document10 pagesKishimoto2021 2022Ana Luiza ArrudaNo ratings yet
- Magnetic Filter FunnelDocument2 pagesMagnetic Filter Funnelsagor sagorNo ratings yet
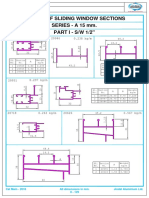
- Sliding Window SectionsDocument52 pagesSliding Window SectionsVSMS8678No ratings yet
- PDS Careclean Bulk CRDocument2 pagesPDS Careclean Bulk CRAdrian FlorinNo ratings yet
- Sea Water DistillationDocument1 pageSea Water DistillationRed one67% (3)
- Books Doubtnut Question BankDocument123 pagesBooks Doubtnut Question Bankasmit6972No ratings yet
- CH - Ash Pond Lining With Geomembrane, NelloreDocument3 pagesCH - Ash Pond Lining With Geomembrane, NellorePa Dadang0% (1)
- Resource Recovery Assessment at A Pulp Mill Wastewater Treatment Plant in UruguayDocument9 pagesResource Recovery Assessment at A Pulp Mill Wastewater Treatment Plant in UruguayElSujeto 999 :3No ratings yet
- Section 2Document60 pagesSection 2Prince EugoNo ratings yet
- Pulp Paper Industry PDFDocument2 pagesPulp Paper Industry PDFX800XLNo ratings yet
- Comprehensive Coordination ChemistryDocument818 pagesComprehensive Coordination Chemistryrace egrNo ratings yet
- Wine Mouthfeel and Texture Author Seth CohenDocument24 pagesWine Mouthfeel and Texture Author Seth CohenMiho CraftsNo ratings yet
- MaretDocument15 pagesMaretslamet siagianNo ratings yet
- XRF-2020 Series: Coating Thickness Gauge Elemental Analysis ROHS AnalyzerDocument3 pagesXRF-2020 Series: Coating Thickness Gauge Elemental Analysis ROHS AnalyzerPhạm DươngNo ratings yet
- GCSE Chemistry Notes: The Chemical Reactions of Common Mineral AcidsDocument10 pagesGCSE Chemistry Notes: The Chemical Reactions of Common Mineral AcidsHanaa AbouziedNo ratings yet
- Method Statement ConbextraGPDocument7 pagesMethod Statement ConbextraGPsinambeladavidNo ratings yet