Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Modeling of Ammonia Removal From Wastewater Using Air Stripping/ Modified Clinoptilolite: Reusability, Optimization, Isotherm, Kinetic, and Equilibrium StudiesDocument22 pagesModeling of Ammonia Removal From Wastewater Using Air Stripping/ Modified Clinoptilolite: Reusability, Optimization, Isotherm, Kinetic, and Equilibrium StudiesvinodNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Final Theses Sunny M.Ed.Document101 pagesFinal Theses Sunny M.Ed.vinodNo ratings yet
- Gokul - 8.7.16Document8 pagesGokul - 8.7.16vinodNo ratings yet
- Water Spray Reactor For Ammonia Removal Via Air Stripping: An Evaluation On Mass Transfer and Process EfficiencyDocument8 pagesWater Spray Reactor For Ammonia Removal Via Air Stripping: An Evaluation On Mass Transfer and Process EfficiencyvinodNo ratings yet
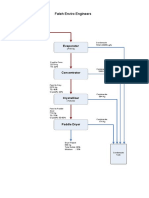
- Fateh Enviro Engineers SchemeDocument1 pageFateh Enviro Engineers SchemevinodNo ratings yet
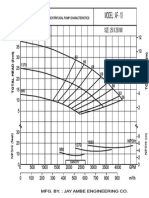
- Jay Ambe AF - 16 CurveDocument1 pageJay Ambe AF - 16 CurvevinodNo ratings yet
- Jay Ambe Axial Pump AF - 10 CurveDocument1 pageJay Ambe Axial Pump AF - 10 CurvevinodNo ratings yet
- Kriloskar KSMB Pump SeriesDocument1 pageKriloskar KSMB Pump SeriesvinodNo ratings yet
- Stripper ColumnDocument1 pageStripper ColumnvinodNo ratings yet
- Operation Characteristic of A Mechanical Vapor Recompression Heat Pump Driven by A Centrifugal FanDocument8 pagesOperation Characteristic of A Mechanical Vapor Recompression Heat Pump Driven by A Centrifugal FanvinodNo ratings yet
- Artificial IntelligenceDocument14 pagesArtificial IntelligencevinodNo ratings yet
- 0.5 MLD PlantDocument1 page0.5 MLD PlantvinodNo ratings yet
- Img 0006-1Document1 pageImg 0006-1vinodNo ratings yet
- PT Mass BalanceDocument3 pagesPT Mass BalancevinodNo ratings yet
- Process Biochemistry: Beatriz Veleirinho, J.A. Lopes-da-SilvaDocument4 pagesProcess Biochemistry: Beatriz Veleirinho, J.A. Lopes-da-SilvavinodNo ratings yet
- Manish KumarDocument4 pagesManish KumarvinodNo ratings yet
- Use of Steam and Co2 As Activating AgentsDocument9 pagesUse of Steam and Co2 As Activating AgentsvinodNo ratings yet
- Chemical For NeutralizationDocument2 pagesChemical For NeutralizationvinodNo ratings yet
- Shylaja 2019Document9 pagesShylaja 2019G G HegdeNo ratings yet
- Concrete Mix Design Mixorder C15-20 - 011743 PDFDocument1 pageConcrete Mix Design Mixorder C15-20 - 011743 PDFkarume ndirituNo ratings yet
- QB 5 - Basic NDT - LT QBDocument5 pagesQB 5 - Basic NDT - LT QBprabhakaran.SNo ratings yet
- Physico-Mechanical Properties and Automotive Fuel Resistance of Epdm-Enr Blends With Hybrid FillersDocument7 pagesPhysico-Mechanical Properties and Automotive Fuel Resistance of Epdm-Enr Blends With Hybrid FillersArjun Satheesh KumarNo ratings yet
- Astm D 3174-04Document5 pagesAstm D 3174-04Servando LozanoNo ratings yet
- TS 1 To 6Document11 pagesTS 1 To 6Anshul Gautam100% (1)
- Liquid CrystalsDocument33 pagesLiquid Crystalsvarundasjh80% (10)
- 5 Accuracy HWDocument2 pages5 Accuracy HWLÂM VŨ THÙYNo ratings yet
- 5.111 Principles of Chemical Science: Mit OpencoursewareDocument6 pages5.111 Principles of Chemical Science: Mit OpencoursewaresarjitgaurNo ratings yet
- Analysis of TriglyceridesDocument8 pagesAnalysis of Triglyceridesdstar13No ratings yet
- Fosroc Nitoflor FC150: Constructive SolutionsDocument4 pagesFosroc Nitoflor FC150: Constructive SolutionsABHI MITRANo ratings yet
- 1 Maxwell's Equations in Matter (Integrate With Next Section)Document2 pages1 Maxwell's Equations in Matter (Integrate With Next Section)iordache100% (1)
- 17 97Document1 page17 97Luis EstradaNo ratings yet
- General Chapters - 711 - DISSOLUTIONDocument11 pagesGeneral Chapters - 711 - DISSOLUTIONFitri WahyuningsihNo ratings yet
- Frt11 BetzDocument25 pagesFrt11 BetzArindam DasNo ratings yet
- 9701 s07 QP 2Document12 pages9701 s07 QP 2Hubbak KhanNo ratings yet
- Magnetism in SSDocument12 pagesMagnetism in SSSusheel WankhedeNo ratings yet
- 6 Single Phase AC Voltage Controller With R and RL LoadsDocument6 pages6 Single Phase AC Voltage Controller With R and RL LoadsSeminars BRECWNo ratings yet
- Gas Sweetening Simulation and Its Optimization by Two Typical AmineDocument8 pagesGas Sweetening Simulation and Its Optimization by Two Typical AmineYogesh PatilNo ratings yet
- Drafting of Heat ExchangerDocument1 pageDrafting of Heat ExchangerHeru YulindoNo ratings yet
- Gagani2017 PDFDocument15 pagesGagani2017 PDFGautamNo ratings yet
- Wastewater DN80 To DN600 PDFDocument36 pagesWastewater DN80 To DN600 PDFMR seaNo ratings yet
- 0 Material Safety Data Sheet: Benzene MSDSDocument6 pages0 Material Safety Data Sheet: Benzene MSDSAli RazaNo ratings yet
- Lee's DiscDocument3 pagesLee's DiscRocker RanjithNo ratings yet
- REFFIPLANT Training CourseDocument76 pagesREFFIPLANT Training CourseKESAVARAPU UMA SAI MAHESHNo ratings yet
- Chemical Bonding 05 Class Notes PDFDocument19 pagesChemical Bonding 05 Class Notes PDFmodel photo copyNo ratings yet
- HMT16 MarksDocument12 pagesHMT16 MarkstagoreboopathyNo ratings yet
- Equation of State of TitaniumDocument6 pagesEquation of State of TitaniumKing OfheartsNo ratings yet
- Fresh and Hardened Properties of 3D Printable - Paul2018Document9 pagesFresh and Hardened Properties of 3D Printable - Paul2018AliReza ZiaratiNo ratings yet