Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5825)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (903)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- HW 1Document2 pagesHW 1Sambhav Jain0% (2)
- TOS 1-Determinacy and StabilityDocument3 pagesTOS 1-Determinacy and StabilityAndrei AlidoNo ratings yet
- Characterization of Models For Time-Dependent Behavior of SoilsDocument21 pagesCharacterization of Models For Time-Dependent Behavior of SoilsrkNo ratings yet
- Why Jacketed Piping Is UsedDocument3 pagesWhy Jacketed Piping Is UsedPranav PatelNo ratings yet
- Lec 12 SchroeEqDocument26 pagesLec 12 SchroeEqKing of KingsNo ratings yet
- Measuring Temperature - Planck's LawDocument2 pagesMeasuring Temperature - Planck's LawNikunj PipariyaNo ratings yet
- Ray OpticsDocument81 pagesRay OpticsGoku HanNo ratings yet
- Energy Transport in A WaveDocument28 pagesEnergy Transport in A WaveAnindya BiswasNo ratings yet
- Salutogenesis IIDocument22 pagesSalutogenesis IIsensorseekerNo ratings yet
- Beer Lambert LawDocument2 pagesBeer Lambert LawMuhammad ZubairNo ratings yet
- Realist AnalysisDocument19 pagesRealist AnalysisLe Thu ThaoNo ratings yet
- Department of Mechanical Engineering: List of Experiments Hvac LabDocument3 pagesDepartment of Mechanical Engineering: List of Experiments Hvac LabHamid MasoodNo ratings yet
- L-2/T-L/Eeef.: .Tul - I. - CB) - .Document44 pagesL-2/T-L/Eeef.: .Tul - I. - CB) - .MeowNo ratings yet
- Assignment Solution 7Document10 pagesAssignment Solution 7nzb1234No ratings yet
- Liquid CrystalsDocument33 pagesLiquid Crystalsvarundasjh80% (10)
- Mechatronic Engineering Technology - K22Document26 pagesMechatronic Engineering Technology - K22dtheanh34No ratings yet
- Current Harmonic Suppression For Dual Three-Phase PDFDocument11 pagesCurrent Harmonic Suppression For Dual Three-Phase PDFMoyasserengAlattarNo ratings yet
- CH11 2Document20 pagesCH11 2Usaid KhanNo ratings yet
- SAMPLE PAPER Physics XI by S.K. Pandey Principal KV GangraniDocument9 pagesSAMPLE PAPER Physics XI by S.K. Pandey Principal KV GangraniAKASH KUMAR X ANo ratings yet
- Influence of Impeller Geometry On HydromechanicalDocument48 pagesInfluence of Impeller Geometry On HydromechanicalNdvs PrasadNo ratings yet
- EN - 840D SL - 5-Axis Training Manual - v26Document388 pagesEN - 840D SL - 5-Axis Training Manual - v26emir_delic2810No ratings yet
- 30072-001-48J (E) .fm5 Page 1 Wednesday, June 23, 1999 4:04 PMDocument3 pages30072-001-48J (E) .fm5 Page 1 Wednesday, June 23, 1999 4:04 PMJose Miguel Cujilema CujilemaNo ratings yet
- Curie Temp of FerroelectricsDocument8 pagesCurie Temp of FerroelectricskanchankonwarNo ratings yet
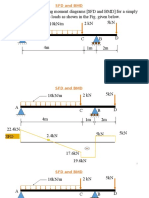
- SFD and BMDDocument6 pagesSFD and BMDHimanshu PathakNo ratings yet
- A Comparative Analysis of Piezoelectric Bending-Mode ActuatorsDocument11 pagesA Comparative Analysis of Piezoelectric Bending-Mode Actuatorsshivgupta2188No ratings yet
- Research Paper On BearingsDocument9 pagesResearch Paper On BearingsmahakNo ratings yet
- Introduction To Openqbmm and Quadrature-Based Moment MethodsDocument53 pagesIntroduction To Openqbmm and Quadrature-Based Moment MethodsMohamed BoukhelefNo ratings yet
- Calorimetry: Heat of SolutionDocument15 pagesCalorimetry: Heat of SolutionsofiaNo ratings yet
- MA8353-Transforms and Partial Differential EquationsDocument17 pagesMA8353-Transforms and Partial Differential Equationssathesh waranNo ratings yet
- 12 Physics Notes Ch05 Magnetism and MatterDocument2 pages12 Physics Notes Ch05 Magnetism and MatterRishabhNo ratings yet