
NFKB3
NFKB3
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5823)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Bequia - Reverse Osmosis Water Treatment System System Description PDFDocument44 pagesBequia - Reverse Osmosis Water Treatment System System Description PDFLiney Gutiérrez Orozco0% (1)
- HUATONG ESP CableDocument18 pagesHUATONG ESP CableSIMON S. FLORES G.100% (1)
- Method Statement For Installation of Water Spray Deluge System - LPG TankDocument13 pagesMethod Statement For Installation of Water Spray Deluge System - LPG Tankpunk cm100% (2)
- FLEXIBLE FLOWLINE RELOCATION & TIE-IN PROCEDURE Rev A - 12022-AMC-TIN-PRO-0016 - IssuedDocument94 pagesFLEXIBLE FLOWLINE RELOCATION & TIE-IN PROCEDURE Rev A - 12022-AMC-TIN-PRO-0016 - IssuedWilliam O Okolotu100% (1)
- Reviews: Micrornas in Myocardial InfarctionDocument8 pagesReviews: Micrornas in Myocardial InfarctionAriadna Zamora GuevaraNo ratings yet
- Reviews: Long Noncoding Rnas in Cardiac Development and AgeingDocument11 pagesReviews: Long Noncoding Rnas in Cardiac Development and AgeingAriadna Zamora GuevaraNo ratings yet
- Oral Maxillary Exostosis: Images in Clinical MedicineDocument1 pageOral Maxillary Exostosis: Images in Clinical MedicineAriadna Zamora GuevaraNo ratings yet
- GraficasDocument5 pagesGraficasAriadna Zamora GuevaraNo ratings yet
- Instalatii Electrice AntiexDocument226 pagesInstalatii Electrice AntiexTibi0% (1)
- Advertisement: Read The Advertisement Below and Answer Questions 3 To 4Document5 pagesAdvertisement: Read The Advertisement Below and Answer Questions 3 To 4Fix Strong VerryNo ratings yet
- The Digestive System EssayDocument3 pagesThe Digestive System EssayJerilee SoCute Watts0% (1)
- Prolog Variabilitas: Apt. Ika Mustikaningtias, M.Sc. Lab Farmakologi Dan Farmasi Klinik Jurusan Farmasi FIKES UNSOEDDocument11 pagesProlog Variabilitas: Apt. Ika Mustikaningtias, M.Sc. Lab Farmakologi Dan Farmasi Klinik Jurusan Farmasi FIKES UNSOEDdhanaNo ratings yet
- Quality Bydesign Principlesfor The Evaluation of Empty Hard CapsulesDocument8 pagesQuality Bydesign Principlesfor The Evaluation of Empty Hard CapsulesNabla ciencia en movimientoNo ratings yet
- Meals Lesson 1Document5 pagesMeals Lesson 1Agagwa Agagwa100% (1)
- Formula Book - Cement IndustriesDocument90 pagesFormula Book - Cement IndustriesKhaled Elemam100% (13)
- SPM Choke Valves Operation Instruction and Service ManualDocument37 pagesSPM Choke Valves Operation Instruction and Service Manualbill100% (1)
- PieEconomics - Cavitation Radiation ReplicationDocument26 pagesPieEconomics - Cavitation Radiation ReplicationshizuyeNo ratings yet
- AIRPHIL V PenswellDocument11 pagesAIRPHIL V PenswellAnonymous fnlSh4KHIgNo ratings yet
- Review On Shilajit Used in Traditional Indian Medicine: Journal of EthnopharmacologyDocument9 pagesReview On Shilajit Used in Traditional Indian Medicine: Journal of EthnopharmacologyDeeksha Baliyan MalikNo ratings yet
- Rietveld Refinement From Powder Diffraction DataDocument47 pagesRietveld Refinement From Powder Diffraction DatamunaphysicsNo ratings yet
- Reading Comprehension SkillDocument25 pagesReading Comprehension SkillFadilNo ratings yet
- Green Chemical ReactionsDocument241 pagesGreen Chemical ReactionsDaiane SantanaNo ratings yet
- Koleston Perfect Fact SheetDocument2 pagesKoleston Perfect Fact SheetMadalina PanduruNo ratings yet
- Pop Brixton + Flimwell Woodland Enterprise Centre - Tech Joint Group - Year 2Document15 pagesPop Brixton + Flimwell Woodland Enterprise Centre - Tech Joint Group - Year 2Mário Alcântara MonteiroNo ratings yet
- Elastomeric Bearings in Polychloroprene VS Natural RubberDocument4 pagesElastomeric Bearings in Polychloroprene VS Natural RubberMiguel Belda DiezNo ratings yet
- Tylenol (Acetaminophen) Dosing, Indications, Interactions, Adverse Effects, and MoreDocument4 pagesTylenol (Acetaminophen) Dosing, Indications, Interactions, Adverse Effects, and MoreMas BroNo ratings yet
- Biology Form 4 Chapter 5 Cell DivisionDocument7 pagesBiology Form 4 Chapter 5 Cell Divisionizuaf817No ratings yet
- Organic Chemistry Tulane SyllabusDocument6 pagesOrganic Chemistry Tulane SyllabusTeehee JonesNo ratings yet
- Aggregation of Charged Colloidal ParticlesDocument41 pagesAggregation of Charged Colloidal ParticlesThu Thảo TrầnNo ratings yet
- Decontamination Techniques Used in Decommissioning ActivitiesDocument51 pagesDecontamination Techniques Used in Decommissioning ActivitiesBiciclistNo ratings yet
- Bright Bar: What We Do, Where To Find UsDocument8 pagesBright Bar: What We Do, Where To Find UsindicomNo ratings yet
- Transparent ElectronicsDocument2 pagesTransparent ElectronicsmanojNo ratings yet
- Electrogravimetry - CHM4112LDocument7 pagesElectrogravimetry - CHM4112Lmarcia1416No ratings yet
- Isomerism DPPDocument4 pagesIsomerism DPPRAGHUL MNo ratings yet
















































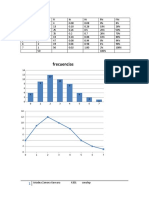


























Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5823)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Bequia - Reverse Osmosis Water Treatment System System Description PDFDocument44 pagesBequia - Reverse Osmosis Water Treatment System System Description PDFLiney Gutiérrez Orozco0% (1)
- HUATONG ESP CableDocument18 pagesHUATONG ESP CableSIMON S. FLORES G.100% (1)
- Method Statement For Installation of Water Spray Deluge System - LPG TankDocument13 pagesMethod Statement For Installation of Water Spray Deluge System - LPG Tankpunk cm100% (2)
- FLEXIBLE FLOWLINE RELOCATION & TIE-IN PROCEDURE Rev A - 12022-AMC-TIN-PRO-0016 - IssuedDocument94 pagesFLEXIBLE FLOWLINE RELOCATION & TIE-IN PROCEDURE Rev A - 12022-AMC-TIN-PRO-0016 - IssuedWilliam O Okolotu100% (1)
- Reviews: Micrornas in Myocardial InfarctionDocument8 pagesReviews: Micrornas in Myocardial InfarctionAriadna Zamora GuevaraNo ratings yet
- Reviews: Long Noncoding Rnas in Cardiac Development and AgeingDocument11 pagesReviews: Long Noncoding Rnas in Cardiac Development and AgeingAriadna Zamora GuevaraNo ratings yet
- Oral Maxillary Exostosis: Images in Clinical MedicineDocument1 pageOral Maxillary Exostosis: Images in Clinical MedicineAriadna Zamora GuevaraNo ratings yet
- GraficasDocument5 pagesGraficasAriadna Zamora GuevaraNo ratings yet
- Instalatii Electrice AntiexDocument226 pagesInstalatii Electrice AntiexTibi0% (1)
- Advertisement: Read The Advertisement Below and Answer Questions 3 To 4Document5 pagesAdvertisement: Read The Advertisement Below and Answer Questions 3 To 4Fix Strong VerryNo ratings yet
- The Digestive System EssayDocument3 pagesThe Digestive System EssayJerilee SoCute Watts0% (1)
- Prolog Variabilitas: Apt. Ika Mustikaningtias, M.Sc. Lab Farmakologi Dan Farmasi Klinik Jurusan Farmasi FIKES UNSOEDDocument11 pagesProlog Variabilitas: Apt. Ika Mustikaningtias, M.Sc. Lab Farmakologi Dan Farmasi Klinik Jurusan Farmasi FIKES UNSOEDdhanaNo ratings yet
- Quality Bydesign Principlesfor The Evaluation of Empty Hard CapsulesDocument8 pagesQuality Bydesign Principlesfor The Evaluation of Empty Hard CapsulesNabla ciencia en movimientoNo ratings yet
- Meals Lesson 1Document5 pagesMeals Lesson 1Agagwa Agagwa100% (1)
- Formula Book - Cement IndustriesDocument90 pagesFormula Book - Cement IndustriesKhaled Elemam100% (13)
- SPM Choke Valves Operation Instruction and Service ManualDocument37 pagesSPM Choke Valves Operation Instruction and Service Manualbill100% (1)
- PieEconomics - Cavitation Radiation ReplicationDocument26 pagesPieEconomics - Cavitation Radiation ReplicationshizuyeNo ratings yet
- AIRPHIL V PenswellDocument11 pagesAIRPHIL V PenswellAnonymous fnlSh4KHIgNo ratings yet
- Review On Shilajit Used in Traditional Indian Medicine: Journal of EthnopharmacologyDocument9 pagesReview On Shilajit Used in Traditional Indian Medicine: Journal of EthnopharmacologyDeeksha Baliyan MalikNo ratings yet
- Rietveld Refinement From Powder Diffraction DataDocument47 pagesRietveld Refinement From Powder Diffraction DatamunaphysicsNo ratings yet
- Reading Comprehension SkillDocument25 pagesReading Comprehension SkillFadilNo ratings yet
- Green Chemical ReactionsDocument241 pagesGreen Chemical ReactionsDaiane SantanaNo ratings yet
- Koleston Perfect Fact SheetDocument2 pagesKoleston Perfect Fact SheetMadalina PanduruNo ratings yet
- Pop Brixton + Flimwell Woodland Enterprise Centre - Tech Joint Group - Year 2Document15 pagesPop Brixton + Flimwell Woodland Enterprise Centre - Tech Joint Group - Year 2Mário Alcântara MonteiroNo ratings yet
- Elastomeric Bearings in Polychloroprene VS Natural RubberDocument4 pagesElastomeric Bearings in Polychloroprene VS Natural RubberMiguel Belda DiezNo ratings yet
- Tylenol (Acetaminophen) Dosing, Indications, Interactions, Adverse Effects, and MoreDocument4 pagesTylenol (Acetaminophen) Dosing, Indications, Interactions, Adverse Effects, and MoreMas BroNo ratings yet
- Biology Form 4 Chapter 5 Cell DivisionDocument7 pagesBiology Form 4 Chapter 5 Cell Divisionizuaf817No ratings yet
- Organic Chemistry Tulane SyllabusDocument6 pagesOrganic Chemistry Tulane SyllabusTeehee JonesNo ratings yet
- Aggregation of Charged Colloidal ParticlesDocument41 pagesAggregation of Charged Colloidal ParticlesThu Thảo TrầnNo ratings yet
- Decontamination Techniques Used in Decommissioning ActivitiesDocument51 pagesDecontamination Techniques Used in Decommissioning ActivitiesBiciclistNo ratings yet
- Bright Bar: What We Do, Where To Find UsDocument8 pagesBright Bar: What We Do, Where To Find UsindicomNo ratings yet
- Transparent ElectronicsDocument2 pagesTransparent ElectronicsmanojNo ratings yet
- Electrogravimetry - CHM4112LDocument7 pagesElectrogravimetry - CHM4112Lmarcia1416No ratings yet
- Isomerism DPPDocument4 pagesIsomerism DPPRAGHUL MNo ratings yet