Download as docx, pdf, or txt
You might also like
- Book - Orthomolecular Psychiatry, Dawkins and Pauling 1973 PDFDocument92 pagesBook - Orthomolecular Psychiatry, Dawkins and Pauling 1973 PDFMarco Cosentino100% (1)
- Newborn Exam Checklist: Examination Osce Items Inspection Vital SignsDocument2 pagesNewborn Exam Checklist: Examination Osce Items Inspection Vital SignsWajid Hussein0% (1)
- Halil İnalcık - The Ottoman Economic Mind and Aspects of The Ottoman Economy PDFDocument10 pagesHalil İnalcık - The Ottoman Economic Mind and Aspects of The Ottoman Economy PDFA Seven HasdemirNo ratings yet
- Welding SymbolsDocument60 pagesWelding Symbolsmitrasah50% (2)
- Fiber Optic Cables and Installation StandardDocument72 pagesFiber Optic Cables and Installation StandardAmol Patki33% (3)
- Comprehensive Pharmacology Study PackageDocument120 pagesComprehensive Pharmacology Study Packageninja-2001No ratings yet
- Drug - Drug Int2Document35 pagesDrug - Drug Int2alishba100% (2)
- Assignment PharmaDocument10 pagesAssignment Pharmac2bmqsfkp7No ratings yet
- AntiepilepticsDocument13 pagesAntiepilepticstbuyinza21apNo ratings yet
- Drug InteractionsDocument30 pagesDrug InteractionsmikhaelyosiaNo ratings yet
- Drug Interactions: MSC Tid 1 Nyakundi BM April 21 2010Document43 pagesDrug Interactions: MSC Tid 1 Nyakundi BM April 21 2010MONALISA MISTRINo ratings yet
- Cclec Drug MonitoringDocument14 pagesCclec Drug MonitoringAndrea Gail GutierrezNo ratings yet
- Bioavailability and Bioequivalence. Therapeutic Drug MonitoringDocument3 pagesBioavailability and Bioequivalence. Therapeutic Drug MonitoringJoel MathewNo ratings yet
- Proses F.kinetik-F.dinamik Ppds 2017Document33 pagesProses F.kinetik-F.dinamik Ppds 2017intan purnamaNo ratings yet
- L5 Drug Action and ToxicityDocument26 pagesL5 Drug Action and Toxicitymurphy 1087No ratings yet
- Clinical PharmacologDocument81 pagesClinical PharmacologSHILOTANo ratings yet
- Pharmacodynamics3 161031154812Document43 pagesPharmacodynamics3 161031154812Adeel Khan100% (1)
- Drug InteractionsDocument43 pagesDrug InteractionsNinaNo ratings yet
- Pharmacology Study NotesDocument121 pagesPharmacology Study Notesninja-2001100% (1)
- Rumana Shaikh MA333 Pharmacology Final ControlDocument6 pagesRumana Shaikh MA333 Pharmacology Final ControlRumana ShaikhNo ratings yet
- Interaksi Obat BukuDocument147 pagesInteraksi Obat BukuSandhy TampubolonNo ratings yet
- Fate of The Drug: Joanne Katherine T. Manlusoc, MSCDocument29 pagesFate of The Drug: Joanne Katherine T. Manlusoc, MSCChinenye AkwueNo ratings yet
- Drug InteractionsDocument21 pagesDrug InteractionsPawan Deshmukh100% (1)
- 41-46 TEXTBOOK The Asam Essentials of Addiction MedicineDocument7 pages41-46 TEXTBOOK The Asam Essentials of Addiction MedicineErlanNo ratings yet
- PharmacologyDocument54 pagesPharmacologyAngelica Palac100% (1)
- PHARMACOLOGYDocument12 pagesPHARMACOLOGYhassangarba6464No ratings yet
- Lecture 2 - Basic Concepts of PharmacokineticsDocument63 pagesLecture 2 - Basic Concepts of Pharmacokineticsahmadslayman1No ratings yet
- Factors Modifying Drug EffectDocument43 pagesFactors Modifying Drug EffectSunil100% (4)
- SummaryDocument7 pagesSummaryMj KhNo ratings yet
- RANASINGHE Basic Pharmacology and IV HypnoticsDocument34 pagesRANASINGHE Basic Pharmacology and IV HypnoticsDagimNo ratings yet
- Principles of Basic Clinical Pharmacokinetic ParametersDocument7 pagesPrinciples of Basic Clinical Pharmacokinetic ParametersThe Ghost By HarethNo ratings yet
- CU SHAH - Refresher Course - 02-10-2016 FinalDocument38 pagesCU SHAH - Refresher Course - 02-10-2016 FinalParthMairNo ratings yet
- Materi 3 - Sifat FisikokimiaDocument61 pagesMateri 3 - Sifat Fisikokimiaashley vechtersbaasNo ratings yet
- PHARMACODYNAMICSDocument62 pagesPHARMACODYNAMICSJulius Kent QuilapioNo ratings yet
- PharmaceuticsDocument20 pagesPharmaceuticsSwastika BastolaNo ratings yet
- PK, PD and Fate of Drug-1Document59 pagesPK, PD and Fate of Drug-1Kenny NgowiNo ratings yet
- General PharmacologyDocument22 pagesGeneral Pharmacologytarek ahmedNo ratings yet
- Drug Interaction Can Be Defined As TheDocument34 pagesDrug Interaction Can Be Defined As TheIstianah Es100% (1)
- Revision KitDocument58 pagesRevision KitTracy MNo ratings yet
- Ceutics CAT 2Document9 pagesCeutics CAT 2Ria KitlerNo ratings yet
- Pharmacokinetics: DR Narendra KumarDocument60 pagesPharmacokinetics: DR Narendra Kumarperala vinaykumarNo ratings yet
- Phar 2Document54 pagesPhar 2Angelica PalacNo ratings yet
- Drug InteractionsDocument5 pagesDrug Interactionsvajkember100% (1)
- 03 - Principles of PharmacokineticsDocument25 pages03 - Principles of PharmacokineticsSimonNo ratings yet
- 1-Lecture One IntroductionDocument45 pages1-Lecture One IntroductionKerolus Joseph AminNo ratings yet
- Share Pharmacology Book 1Document101 pagesShare Pharmacology Book 1Jmcle AhmedNo ratings yet
- Pharmacodynamics, PharmacokineticsDocument7 pagesPharmacodynamics, PharmacokineticsSammee RegoraNo ratings yet
- Pharmacology Unit 1Document58 pagesPharmacology Unit 1Asif Ali LashariNo ratings yet
- Drug InteractionsDocument27 pagesDrug InteractionsodiwuorvicksonNo ratings yet
- Biofar&farmakokinetikDocument34 pagesBiofar&farmakokinetikYayuk Abay TambunanNo ratings yet
- Drug Interactions: DR - Omer A.A.S. Rikaby BSC Pharmacy MSC Clinical Pharmacy MbaDocument40 pagesDrug Interactions: DR - Omer A.A.S. Rikaby BSC Pharmacy MSC Clinical Pharmacy MbaAbdalla Kazzam100% (1)
- PHARMACOKINETICS & PharmacokineticsDocument60 pagesPHARMACOKINETICS & PharmacokineticsRaj PathakNo ratings yet
- PK, PD, Drug Interaction and Toxicology Bmls3 2020 2Document189 pagesPK, PD, Drug Interaction and Toxicology Bmls3 2020 2Kenny NgowiNo ratings yet
- Physicochemical Properties SksDocument61 pagesPhysicochemical Properties SksGokul Raj.PNo ratings yet
- The Ladmer SystemDocument12 pagesThe Ladmer Systemkriss WongNo ratings yet
- Drug InteractionDocument27 pagesDrug InteractionRida Nur RafidahNo ratings yet
- Lecture-13 Drug SpecificityDocument15 pagesLecture-13 Drug SpecificityChian WrightNo ratings yet
- Pharmacodyanamics-Model Questions & AnswersDocument6 pagesPharmacodyanamics-Model Questions & AnswersDr.U.P.Rathnakar.MD.DIH.PGDHMNo ratings yet
- Variability in Drug ResponseDocument18 pagesVariability in Drug Responsehlouis8No ratings yet
- Per 1 - 4 Farmakologi UmumDocument98 pagesPer 1 - 4 Farmakologi UmumamaliahriskaikaNo ratings yet
- Handbook of Drug Interaction and the Mechanism of InteractionFrom EverandHandbook of Drug Interaction and the Mechanism of InteractionRating: 1 out of 5 stars1/5 (1)
- An Introduction to Mechanisms in Pharmacology and TherapeuticsFrom EverandAn Introduction to Mechanisms in Pharmacology and TherapeuticsNo ratings yet
- Chapter 17: Study Documentation: Summary of Edits Between Version 4.0 and 5.0Document8 pagesChapter 17: Study Documentation: Summary of Edits Between Version 4.0 and 5.0Wajid HusseinNo ratings yet
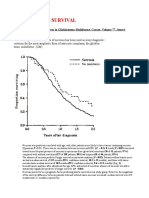
- Necrosis and Survival: Necrosis As A Prognostic Factor in Glioblastoma MultiformeDocument2 pagesNecrosis and Survival: Necrosis As A Prognostic Factor in Glioblastoma MultiformeWajid HusseinNo ratings yet
- Improving Memory and Study Skills: Advances in Theory and PracticeDocument14 pagesImproving Memory and Study Skills: Advances in Theory and PracticeWajid HusseinNo ratings yet
- Mac OS X Keyboard CommandsDocument9 pagesMac OS X Keyboard CommandsWajid HusseinNo ratings yet
- Full Blood CountDocument4 pagesFull Blood CountWajid HusseinNo ratings yet
- Format For Clerking Neonates CHeRP 2007Document1 pageFormat For Clerking Neonates CHeRP 2007Wajid HusseinNo ratings yet
- p61 PDFDocument8 pagesp61 PDFWajid HusseinNo ratings yet
- Pizza Modo MioDocument20 pagesPizza Modo MioLucas Horacio0% (1)
- This Study Resource Was: Asynchronous Activity: Asean Literature April 21, 2021, WednesdayDocument2 pagesThis Study Resource Was: Asynchronous Activity: Asean Literature April 21, 2021, WednesdayPatricia ByunNo ratings yet
- 2ND Quiz Eng3aDocument3 pages2ND Quiz Eng3aDiona MacasaquitNo ratings yet
- Lop 10 On Thi Giua Ki. Buoi 2. in Class 2Document8 pagesLop 10 On Thi Giua Ki. Buoi 2. in Class 2Nguyễn AnNo ratings yet
- Ktda Top Average Price and Sold Quantities Sale 20 - 240403 - 175941Document2 pagesKtda Top Average Price and Sold Quantities Sale 20 - 240403 - 175941InnocentNo ratings yet
- LP - Graphic MaterialsDocument2 pagesLP - Graphic MaterialsRuby Flor Dela CruzNo ratings yet
- Pathology Revision.: EmailDocument4 pagesPathology Revision.: EmailRiyaduNo ratings yet
- Uts SyntaxDocument2 pagesUts SyntaxFerdiansyahNo ratings yet
- Transport Lesson PlanDocument4 pagesTransport Lesson PlanKapten HanzwiraNo ratings yet
- Buddhism Lamp PDFDocument193 pagesBuddhism Lamp PDFrajeshteeNo ratings yet
- Iligan Medical Center CollegeDocument14 pagesIligan Medical Center CollegeMyk Twentytwenty NBeyondNo ratings yet
- Squaring The Range Trading SystemDocument104 pagesSquaring The Range Trading Systemfulgence1No ratings yet
- Year 6 Daily Lesson Plans Success Criteria Pupils Can 1. Guess The Meanings of at Least 5 Words With Suitable Answers. 2. Read and Answer at Least 5 Questions CorrectlyDocument5 pagesYear 6 Daily Lesson Plans Success Criteria Pupils Can 1. Guess The Meanings of at Least 5 Words With Suitable Answers. 2. Read and Answer at Least 5 Questions CorrectlySelva RajaNo ratings yet
- BBM203 - Kierrtana - 071190094Document11 pagesBBM203 - Kierrtana - 071190094kierrtanaNo ratings yet
- Glee.s05e01.Hdtv.x264 LolDocument71 pagesGlee.s05e01.Hdtv.x264 LolraffiisahNo ratings yet
- Testimony of A Former HinduDocument3 pagesTestimony of A Former HinduMarie SantoroNo ratings yet
- Dr. B.R. Ambedkar Open University Examination Branch: NotificationDocument4 pagesDr. B.R. Ambedkar Open University Examination Branch: NotificationKranti KumarNo ratings yet
- GGI and MFR Theory: Master Master Index Help On HelpDocument6 pagesGGI and MFR Theory: Master Master Index Help On HelpIvan TroninNo ratings yet
- 0 Elsevier Scientific Publishing Company,: Amsterdam - Printed inDocument24 pages0 Elsevier Scientific Publishing Company,: Amsterdam - Printed inJulian I SwandiNo ratings yet
- Clinical Biomechanics: Leigh W. Marshall, Stuart M. McgillDocument4 pagesClinical Biomechanics: Leigh W. Marshall, Stuart M. McgillMichael JunNo ratings yet
- Notes-Maternal Health NursingDocument22 pagesNotes-Maternal Health NursingRachael Crossgrove100% (4)
- Mcguire eDocument1 pageMcguire efranjuranNo ratings yet
- An Introduction To Neuroendocrinology (PDFDrive) (1) - Compressed-83-87Document5 pagesAn Introduction To Neuroendocrinology (PDFDrive) (1) - Compressed-83-87lirNo ratings yet
- DVB 043 HuttlDocument9 pagesDVB 043 HuttlStarLink1No ratings yet
- Handout Exercise Inter First Conditional Vs Second Conditional Ex1Document2 pagesHandout Exercise Inter First Conditional Vs Second Conditional Ex1Ljiljana MaksimovicNo ratings yet
- Rauzatal AuliyaDocument113 pagesRauzatal AuliyaMohammed Abdul Hafeez, B.Com., Hyderabad, IndiaNo ratings yet