Download as pdf or txt
You might also like
- Soap Investigatory ProjectDocument28 pagesSoap Investigatory ProjectXyrah Rabanillo67% (3)
- An Introduction to X-Ray Physics, Optics, and ApplicationsFrom EverandAn Introduction to X-Ray Physics, Optics, and ApplicationsRating: 5 out of 5 stars5/5 (1)
- Magnesium Fluoride mgf2 Transmission Curve DatasheetDocument1 pageMagnesium Fluoride mgf2 Transmission Curve DatasheetJong Rok AhnNo ratings yet
- Lek SellDocument51 pagesLek SellgerrymikepalisocNo ratings yet
- Product Sheet ALON PDFDocument3 pagesProduct Sheet ALON PDFMauricio FriedrichNo ratings yet
- Barnes Layer Analysis: R 2par, Per Astm G 57 & Barnes MethodDocument33 pagesBarnes Layer Analysis: R 2par, Per Astm G 57 & Barnes MethodAnonymous lv8SNRyNo ratings yet
- Sky Radiance and ConversionDocument10 pagesSky Radiance and Conversioncours2thomasNo ratings yet
- Progress - AMS Study ErbelDocument19 pagesProgress - AMS Study ErbelmarksteckelNo ratings yet
- Oncor Ag Spec Datasheet-0609Document20 pagesOncor Ag Spec Datasheet-0609jcdavidm2000No ratings yet
- Копия Force, Motion, And Energy - Science - 11th Grade by SlidesgoDocument12 pagesКопия Force, Motion, And Energy - Science - 11th Grade by SlidesgoYasmin GamesNo ratings yet
- 3.5 Core Al Xlpe ArmdDocument1 page3.5 Core Al Xlpe ArmdRavindra JadhavNo ratings yet
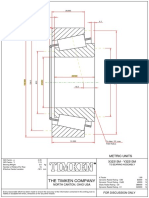
- The Timken Company: North Canton, Ohio UsaDocument1 pageThe Timken Company: North Canton, Ohio UsaChristian HuamanNo ratings yet
- Kolesterol (Kat) X Usia, Imt, LP, RLPTB (Numerik) T-Test: Group StatisticsDocument2 pagesKolesterol (Kat) X Usia, Imt, LP, RLPTB (Numerik) T-Test: Group StatisticsFrancisca NovelianiNo ratings yet
- Kolesterol (Kat) X Usia, Imt, LP, RLPTB (Numerik) T-Test: Group StatisticsDocument2 pagesKolesterol (Kat) X Usia, Imt, LP, RLPTB (Numerik) T-Test: Group StatisticsFrancisca NovelianiNo ratings yet
- Barnes Layer Analysis: R 2par, Per Astm G 57 & Barnes MethodDocument5 pagesBarnes Layer Analysis: R 2par, Per Astm G 57 & Barnes MethodAnonymous lv8SNRyNo ratings yet
- Laboratory Rock Mechanics 6 PDFDocument7 pagesLaboratory Rock Mechanics 6 PDFirsanNo ratings yet
- SESAM Saturable AbsorbersDocument51 pagesSESAM Saturable AbsorbersVineeth PLNo ratings yet
- CSections LoadTable-ExDocument8 pagesCSections LoadTable-Exعبد الباسط المغيربيNo ratings yet
- RS603MDocument3 pagesRS603Melkin_oviedoNo ratings yet
- RPM Dial Reading True Yield Taza de Corte (Y) Sec-1 Shear Stress T (lb/100ft2) Apparent Viscosity CPDocument38 pagesRPM Dial Reading True Yield Taza de Corte (Y) Sec-1 Shear Stress T (lb/100ft2) Apparent Viscosity CPErika Tanaka100% (1)
- CCD X-Ray Detectors8656Document2 pagesCCD X-Ray Detectors8656richartinNo ratings yet
- MTC Tubes Conares 2020 Ms XsteelDocument1 pageMTC Tubes Conares 2020 Ms Xsteelfahad.outoftheboxNo ratings yet
- Materials 1 Lab ReportDocument8 pagesMaterials 1 Lab ReportarthurNo ratings yet
- Introductory Research PosterDocument1 pageIntroductory Research PosterJustin BallNo ratings yet
- Cantilever-Si Units Final 022615Document111 pagesCantilever-Si Units Final 022615adma1No ratings yet
- Torsional Vibration Spread SheetDocument14 pagesTorsional Vibration Spread SheetRPDeshNo ratings yet
- Experimental Analysis and Results 4.1 Experimental Analysis 4.1.1 Experimental SetupDocument20 pagesExperimental Analysis and Results 4.1 Experimental Analysis 4.1.1 Experimental SetupFortune FireNo ratings yet
- Diffractometric Debye-Scherrer Patterns of Powder SamplesDocument9 pagesDiffractometric Debye-Scherrer Patterns of Powder SamplesJose GalvanNo ratings yet
- Member Name: P1: 1. General InformationDocument6 pagesMember Name: P1: 1. General Informationvijay kumar yadavNo ratings yet
- High Performance - Grade 90: Pretensioned Spun High Strength Concrete PilesDocument2 pagesHigh Performance - Grade 90: Pretensioned Spun High Strength Concrete PilesPete WongNo ratings yet
- Micra MC1VR01 SpecDocument3 pagesMicra MC1VR01 SpecTrình BiomedicNo ratings yet
- Lecture4-RadPhysNucMed PartcDocument15 pagesLecture4-RadPhysNucMed PartcMSNo ratings yet
- CH9 CromwellDocument9 pagesCH9 CromwellPritamsingh SolankiNo ratings yet
- Blackbody Radiation SpectrumDocument3 pagesBlackbody Radiation SpectrumernaNo ratings yet
- Roughness of Coarse AggregateDocument3 pagesRoughness of Coarse AggregateHamada Shoukry MohammedNo ratings yet
- LIM10 - 1 Tabla 1500x3000 - ReportDocument1 pageLIM10 - 1 Tabla 1500x3000 - ReportMarko MarkovicNo ratings yet
- 2013 12 06 StereolithographyDocument36 pages2013 12 06 StereolithographyprabhjotbhangalNo ratings yet
- Diffractometric Debye-Scherrer Patterns of Powder SamplesDocument11 pagesDiffractometric Debye-Scherrer Patterns of Powder SamplesJose GalvanNo ratings yet
- ALON Data SheetDocument1 pageALON Data SheetTran Manh TungNo ratings yet
- Jac M6SF-3-1Document2 pagesJac M6SF-3-1lorens moraNo ratings yet
- StandardPracticeforAssessingtheDegreeofBandingorOrientationofMicrostructuresbyAutomaticImageAnalysis PDFDocument33 pagesStandardPracticeforAssessingtheDegreeofBandingorOrientationofMicrostructuresbyAutomaticImageAnalysis PDFARUN KUMAR THAKURNo ratings yet
- 325W Bifacial Mono PERC Double Glass ModuleDocument2 pages325W Bifacial Mono PERC Double Glass ModuleJosue Enriquez EguigurenNo ratings yet
- Stocker XRD 324-330Document7 pagesStocker XRD 324-330irem AydoganNo ratings yet
- Hollow Section: Circular: A. Cross Section AnalysisDocument5 pagesHollow Section: Circular: A. Cross Section AnalysisinaNo ratings yet
- Chapter 2. X-Ray Diffraction Analysis: Dr. Nguyen Tuan LoiDocument64 pagesChapter 2. X-Ray Diffraction Analysis: Dr. Nguyen Tuan LoiKỲ ĐỖNo ratings yet
- Final Spesifikasi EKAT SOMATOM CONFIDENCE RTDocument1 pageFinal Spesifikasi EKAT SOMATOM CONFIDENCE RTUlfi YunitaNo ratings yet
- Explore: Case Processing SummaryDocument7 pagesExplore: Case Processing SummaryIrma ZarinaNo ratings yet
- Cu/Pvc/Swa/Pvc (Nyrgby) Cu/Pvc/Sfa/Pvc (Nyfgby) 0.6/1 (1.2) KVDocument3 pagesCu/Pvc/Swa/Pvc (Nyrgby) Cu/Pvc/Sfa/Pvc (Nyfgby) 0.6/1 (1.2) KVAgus Muhammad SolehNo ratings yet
- Transvers Analysis 129iDocument22 pagesTransvers Analysis 129iruchitaNo ratings yet
- Aqib XRD ReportDocument25 pagesAqib XRD ReportMohammad DanishNo ratings yet
- Experiment 4 Report 27-2Document4 pagesExperiment 4 Report 27-2sohaila gaberNo ratings yet
- Barnes Layer Analysis: R 2par, Per Astm G 57 & Barnes MethodDocument13 pagesBarnes Layer Analysis: R 2par, Per Astm G 57 & Barnes MethodAnonymous lv8SNRyNo ratings yet
- Soal Lks 2015Document2 pagesSoal Lks 2015Thomas WijayaNo ratings yet
- Technical Seminar Presentation HarishDocument21 pagesTechnical Seminar Presentation Harishprashanth nNo ratings yet
- Basics of X-Ray Powder DiffractionDocument82 pagesBasics of X-Ray Powder DiffractionJohn Gerald OdhiamboNo ratings yet
- 1summary dll-1 - 1Document1 page1summary dll-1 - 1fajriNo ratings yet
- 1997.metzgar Et AlDocument9 pages1997.metzgar Et AlAbheek BardhanNo ratings yet
- 473 - Salazar - N11 Jose Rocco-Diego SalazarDocument10 pages473 - Salazar - N11 Jose Rocco-Diego SalazarJosé RoccoNo ratings yet
- 6 - Two-Way Slab Example 1Document8 pages6 - Two-Way Slab Example 1Richard PekitpekitNo ratings yet
- X-Ray Determination of Retained Austenite in Steel With Near Random Crystallographic OrientationDocument7 pagesX-Ray Determination of Retained Austenite in Steel With Near Random Crystallographic OrientationNatalie FeriaNo ratings yet
- d1/d Parameter in EPRDocument53 pagesd1/d Parameter in EPRBrandon ArceNo ratings yet
- EPR Studies On Photosintetic BacteriaDocument35 pagesEPR Studies On Photosintetic BacteriaBrandon ArceNo ratings yet
- 2010 ArticleDocument9 pages2010 ArticleBrandon ArceNo ratings yet
- Structural Insights Into The Early Steps of Receptor Transducer Signal Transfer in Archaeal PhototaxisDocument8 pagesStructural Insights Into The Early Steps of Receptor Transducer Signal Transfer in Archaeal PhototaxisBrandon ArceNo ratings yet
- SAD Phasing History Current Impact and Future Opportunities 2016 Archives of Biochemistry and BiophysicsDocument15 pagesSAD Phasing History Current Impact and Future Opportunities 2016 Archives of Biochemistry and BiophysicsBrandon ArceNo ratings yet
- 2003 Nab Er Bio PhysDocument7 pages2003 Nab Er Bio PhysBrandon ArceNo ratings yet
- ESRDocument9 pagesESRBrandon ArceNo ratings yet
- Archives of Biochemistry and Biophysics: Bente VestergaardDocument11 pagesArchives of Biochemistry and Biophysics: Bente VestergaardBrandon ArceNo ratings yet
- TPX Capillary & Holder: Maximizing Your PotentialDocument2 pagesTPX Capillary & Holder: Maximizing Your PotentialBrandon ArceNo ratings yet
- Brief Intro To Crystallography TT 2011-02-09Document19 pagesBrief Intro To Crystallography TT 2011-02-09Brandon ArceNo ratings yet
- M&A of Facebook and WhatsappDocument22 pagesM&A of Facebook and WhatsappPriya Upadhyay50% (2)
- Islamabad Master Planning and ArchitectuDocument289 pagesIslamabad Master Planning and ArchitectuAngela Leonardo100% (1)
- Brahms Alto RhapsodyDocument6 pagesBrahms Alto RhapsodyAine Mulvey100% (1)
- The Problem and Review of Related Literature and StudiesDocument11 pagesThe Problem and Review of Related Literature and StudiesVincent TayagNo ratings yet
- The Ten Commandments - Joseph LewisDocument327 pagesThe Ten Commandments - Joseph Lewisdudhai100% (1)
- RYLA Personality Styles I May Not Be PerfectDocument36 pagesRYLA Personality Styles I May Not Be PerfectBryan KoNo ratings yet
- Lesson Plan GrammarDocument3 pagesLesson Plan Grammar25eya100% (2)
- Documento - MX Solucionario Administracioacuten de Operaciones 12va Edicion Chase Richard JacobsDocument10 pagesDocumento - MX Solucionario Administracioacuten de Operaciones 12va Edicion Chase Richard JacobsAnderson InsuastiNo ratings yet
- The Mark of The Beast - Divine Messages To Maria Divine MercyDocument38 pagesThe Mark of The Beast - Divine Messages To Maria Divine Mercykapf2100% (1)
- Brain ImagingDocument14 pagesBrain ImagingSenal Malaka PremarathnaNo ratings yet
- Restoration of Endodontically Treated Teeth 5pdfDocument17 pagesRestoration of Endodontically Treated Teeth 5pdfAlaa MadmoujNo ratings yet
- Mila - DZ - Univ - Ulesbaa@centre .Bo M Mila - DZ - Univ - @centre .Belhaj T Setif - DZ - @univ .Kourtel FDocument22 pagesMila - DZ - Univ - Ulesbaa@centre .Bo M Mila - DZ - Univ - @centre .Belhaj T Setif - DZ - @univ .Kourtel FBelhadj TarekNo ratings yet
- 04 Case # 3 Uy v. Estate of Vipa FernandezDocument3 pages04 Case # 3 Uy v. Estate of Vipa FernandezDee WhyNo ratings yet
- Tribal Network EffectsDocument4 pagesTribal Network Effectsadmin thehearth0% (1)
- Zul Hijjah, Eidul Adha & Qurbani Parts 1-3Document5 pagesZul Hijjah, Eidul Adha & Qurbani Parts 1-3takwaniaNo ratings yet
- Me Too + Social MediaDocument5 pagesMe Too + Social MediaMimi Kelly100% (1)
- Emejulu - Bassel 2020 The Politics of ExhaustionDocument8 pagesEmejulu - Bassel 2020 The Politics of ExhaustionEliana BatistaNo ratings yet
- LCM Summer Training Project Industrial Relations in BSPDocument35 pagesLCM Summer Training Project Industrial Relations in BSPRavi NegiNo ratings yet
- SALAZAR3Document1 pageSALAZAR3Mark SalazarNo ratings yet
- Secretos Medicina Interna FinalDocument251 pagesSecretos Medicina Interna FinalCentroVeterinarioCrubvet100% (3)
- Drug Study On Emergency DrugsDocument34 pagesDrug Study On Emergency DrugsMei-mei ZhuangNo ratings yet
- Transitional Justice HistoriesDocument28 pagesTransitional Justice HistoriesEve AthanasekouNo ratings yet
- Anxiety: A Self Help GuideDocument32 pagesAnxiety: A Self Help Guideabraham.gallegos100% (1)
- Harley DavidsoncustomerrelationshipDocument7 pagesHarley DavidsoncustomerrelationshipSachin GolherNo ratings yet
- 4073Q2 Specimen StatisticsDocument20 pages4073Q2 Specimen StatisticsdvynmashNo ratings yet
- Nursing Care PlanDocument3 pagesNursing Care PlanBlitz KriegNo ratings yet
- Scandent SolutionsDocument4 pagesScandent Solutionsr.jeyashankar9550No ratings yet
- Community Participation and Empowerment in Primary Education PDFDocument2 pagesCommunity Participation and Empowerment in Primary Education PDFRachelNo ratings yet
- The Effect of Oral Care With Chlorhexidine PDFDocument7 pagesThe Effect of Oral Care With Chlorhexidine PDFALISSON DAYAN CHICA HERRERANo ratings yet