
Cher Mette 1998
Cher Mette 1998
You might also like
- BYUOpticsBook 2013Document344 pagesBYUOpticsBook 2013geopaok165No ratings yet
- Dynamic Response of Fixed Offshore Structures Under Environmental LoadsDocument16 pagesDynamic Response of Fixed Offshore Structures Under Environmental LoadsxautraixxxxxxNo ratings yet
- 01 Main Part1 2Document68 pages01 Main Part1 2kashyapNo ratings yet
- Journal of Alloys and Compounds: ReviewDocument11 pagesJournal of Alloys and Compounds: ReviewTalita SouzaNo ratings yet
- 147-Waves, Modes, CommunicationsDocument147 pages147-Waves, Modes, CommunicationsRuifeng XuNo ratings yet
- 232 EtdDocument92 pages232 Etdjan dildoNo ratings yet
- Research PaperDocument49 pagesResearch Paperben0706No ratings yet
- Ktab Mo3ama9 Fih KolachDocument186 pagesKtab Mo3ama9 Fih KolachMustapha OuldacheNo ratings yet
- Non Imagistic Optics Aop-10-2-4841Document29 pagesNon Imagistic Optics Aop-10-2-4841Danut StanciuNo ratings yet
- Carlos S. Kubrusly (Auth.) - Spectral Theory of Operators On Hilbert Spaces-Birkhäuser Basel (2012) PDFDocument208 pagesCarlos S. Kubrusly (Auth.) - Spectral Theory of Operators On Hilbert Spaces-Birkhäuser Basel (2012) PDFMohamed EL BERKAOUI100% (1)
- Sapt DFT PDFDocument75 pagesSapt DFT PDFAnonymous pa8pSCC15No ratings yet
- Sapt DFT PDFDocument75 pagesSapt DFT PDFAnonymous pa8pSCC15No ratings yet
- BTH2012 OloumiDocument71 pagesBTH2012 OloumiMilton AlvesNo ratings yet
- RMIT University: Metamaterial ResonatorsDocument24 pagesRMIT University: Metamaterial Resonatorsronalme100% (1)
- Said DissertationDocument129 pagesSaid DissertationSaeed AzarNo ratings yet
- Numerical Analysis of A Bisection-Exclusion Method To Find Zeros of Univariate Analytic FunctionsDocument39 pagesNumerical Analysis of A Bisection-Exclusion Method To Find Zeros of Univariate Analytic FunctionsLucas SantosNo ratings yet
- Malhotra Et Al 2016Document17 pagesMalhotra Et Al 2016Abhishek MalhotraNo ratings yet
- MT5 PDFDocument80 pagesMT5 PDFAnonymous 8VIwRGNo ratings yet
- Jiang 2017Document49 pagesJiang 2017Agtc TandayNo ratings yet
- Computational Fluid Dynamics Modelling of Proton Exchange Membrane Fuel CellsDocument29 pagesComputational Fluid Dynamics Modelling of Proton Exchange Membrane Fuel CellsrmvanginkelNo ratings yet
- Fwejk544 9949 FRRDocument93 pagesFwejk544 9949 FRRaicreativeintellectNo ratings yet
- C RauzyDocument162 pagesC Rauzyc_otescuNo ratings yet
- Electromagnetic Metasurfaces: Physics and Applications: Advances in Optics and Photonics June 2019Document101 pagesElectromagnetic Metasurfaces: Physics and Applications: Advances in Optics and Photonics June 2019Jojo NamNo ratings yet
- Electrospinning of Polymer Nanofibers - Effects On Oriented Morphology, Structures and Tensile PropertiesDocument16 pagesElectrospinning of Polymer Nanofibers - Effects On Oriented Morphology, Structures and Tensile PropertiesCamilo A. Suárez BallesterosNo ratings yet
- Investigation of Near-Field Contribution in SBR For Installed Antenna PerformanceDocument80 pagesInvestigation of Near-Field Contribution in SBR For Installed Antenna Performancelaythe_99No ratings yet
- Electric-Magnetic Duality and The Geometric Langlands ProgramDocument231 pagesElectric-Magnetic Duality and The Geometric Langlands ProgramEliacim VelezNo ratings yet
- Welding Methods of PolymerDocument10 pagesWelding Methods of PolymerGOPINATH SNo ratings yet
- 1 s2.0 S1364032113007260 MainDocument19 pages1 s2.0 S1364032113007260 MainkitsomacNo ratings yet
- Dissertation / Doctoral Thesis: Ab-Initio Description of Optical Properties of Semiconductors and NanocrystalsDocument200 pagesDissertation / Doctoral Thesis: Ab-Initio Description of Optical Properties of Semiconductors and NanocrystalsSiabdallahNo ratings yet
- Understanding The Reaction of Nuclear GRDocument25 pagesUnderstanding The Reaction of Nuclear GRSandipGangurdeNo ratings yet
- Spectrochimica Acta Part B: E. Tognoni, G. Cristoforetti, S. Legnaioli, V. PalleschiDocument14 pagesSpectrochimica Acta Part B: E. Tognoni, G. Cristoforetti, S. Legnaioli, V. PalleschibetjodaNo ratings yet
- High-Contrast Gratings For Integrated Optoelectronics: Connie J. Chang-Hasnain and Weijian YangDocument62 pagesHigh-Contrast Gratings For Integrated Optoelectronics: Connie J. Chang-Hasnain and Weijian YangMohit SinhaNo ratings yet
- FerritesobtainedbysolgelDocument42 pagesFerritesobtainedbysolgelTasnuva HumairaNo ratings yet
- Analogue Gravity Living ReviewDocument159 pagesAnalogue Gravity Living ReviewTapas DasNo ratings yet
- 1 s2.0 S0950061818330307 MainDocument15 pages1 s2.0 S0950061818330307 MainAKINMADE OLUWATOSINNo ratings yet
- Production of Carbon Nanostructures in Biochar BioDocument15 pagesProduction of Carbon Nanostructures in Biochar BioJatin Kumar SaxenaNo ratings yet
- Mbithi Nelson Matheka PHD 2022Document88 pagesMbithi Nelson Matheka PHD 2022Jenő MiklayNo ratings yet
- Bi Metallic NPs by Chemical MethodDocument251 pagesBi Metallic NPs by Chemical Methodmuhammad siddiq siddiqNo ratings yet
- Progress in Energy and Combustion Science: L.Y.M. Gicquel, G. Staffelbach, T. PoinsotDocument36 pagesProgress in Energy and Combustion Science: L.Y.M. Gicquel, G. Staffelbach, T. PoinsotMoses DevaprasannaNo ratings yet
- 315 TextDocument144 pages315 TextKevin PascualNo ratings yet
- 2012 ELSEVIER XRD In-Situ TestDocument13 pages2012 ELSEVIER XRD In-Situ Testirem AydoganNo ratings yet
- Full TextDocument190 pagesFull Textbsmalah11alroxnamNo ratings yet
- Study of SILAR Deposited ZnO For InverteDocument61 pagesStudy of SILAR Deposited ZnO For InvertekardesimnaberyaNo ratings yet
- Progress in Energy and Combustion Science: Kehang Cui, Shigeo MaruyamaDocument21 pagesProgress in Energy and Combustion Science: Kehang Cui, Shigeo MaruyamaAli AliNo ratings yet
- Limousin, G Et Al. Sorption Isotherms A Review On Physical Bases, Modeling and MeasurementDocument27 pagesLimousin, G Et Al. Sorption Isotherms A Review On Physical Bases, Modeling and MeasurementbrianNo ratings yet
- PVDF HFP 1Document24 pagesPVDF HFP 1salmanNo ratings yet
- A Comparative Study of Total Direct and Diffuse Solar Irradiance byDocument11 pagesA Comparative Study of Total Direct and Diffuse Solar Irradiance bysasa_22No ratings yet
- Materials Science and Engineering C: ReviewDocument16 pagesMaterials Science and Engineering C: ReviewBeesam Ramesh KumarNo ratings yet
- Quantum Hadrodynamics and Dense Matter ThesisDocument135 pagesQuantum Hadrodynamics and Dense Matter ThesisalexisNo ratings yet
- Collections of Math IIDocument561 pagesCollections of Math IIHenry GarrettNo ratings yet
- Cooper and BistDocument21 pagesCooper and BistDiana PleşcaNo ratings yet
- Scaling Laws and Electron Properties in Hall Effect ThrustersDocument176 pagesScaling Laws and Electron Properties in Hall Effect Thrustersaakash30janNo ratings yet
- Seddon 2017Document74 pagesSeddon 2017Rayhan XhanNo ratings yet
- Trig BookDocument203 pagesTrig BookmayNo ratings yet
- DEREK R.MILLER - Nanoscale Metal Oxide-Based Heterojunctions For Gas Sensing - Review PDFDocument23 pagesDEREK R.MILLER - Nanoscale Metal Oxide-Based Heterojunctions For Gas Sensing - Review PDFAna Maria NiculescuNo ratings yet
- Journal of Materiomics: Michael Green, Xiaobo ChenDocument39 pagesJournal of Materiomics: Michael Green, Xiaobo ChenSohail AhmedNo ratings yet
- Silicon 2Document221 pagesSilicon 2cafierroNo ratings yet
- Organic and Perovskite Solar Cells Working Principles, Materials and Interfaces - Marinova - 2016Document17 pagesOrganic and Perovskite Solar Cells Working Principles, Materials and Interfaces - Marinova - 2016Aniello LangellaNo ratings yet
- ICTAC Kinetics Committee Recommendations For Performing Kinetic Computations On Thermal Analysis DataDocument19 pagesICTAC Kinetics Committee Recommendations For Performing Kinetic Computations On Thermal Analysis Datatotenkopf0424No ratings yet
- The Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionFrom EverandThe Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionNo ratings yet
- The Action Principle and Partial Differential Equations. (AM-146), Volume 146From EverandThe Action Principle and Partial Differential Equations. (AM-146), Volume 146No ratings yet
- Patil2012 PDFDocument4 pagesPatil2012 PDFAndrés Camilo LópezNo ratings yet
- Study On The Concrete in Chloride Environment Based On Electrochemical Impedance SpectrosDocument13 pagesStudy On The Concrete in Chloride Environment Based On Electrochemical Impedance SpectrosAndrés Camilo LópezNo ratings yet
- Review ArticleDocument20 pagesReview ArticleAndrés Camilo LópezNo ratings yet
- Pu 2005Document6 pagesPu 2005Andrés Camilo LópezNo ratings yet
- Latex Template For Preparing A Research Article For Submission To The Journal of Optical Communications and NetworkingDocument3 pagesLatex Template For Preparing A Research Article For Submission To The Journal of Optical Communications and NetworkingAndrés Camilo LópezNo ratings yet
- Global Hybrid Functionals: A Look at The Engine Under The HoodDocument16 pagesGlobal Hybrid Functionals: A Look at The Engine Under The HoodAndrés Camilo LópezNo ratings yet
- Does A Photochemical Reaction Have A Reaction Order?: S. R. LoganDocument1 pageDoes A Photochemical Reaction Have A Reaction Order?: S. R. LoganAndrés Camilo LópezNo ratings yet
- Lec2 PDFDocument4 pagesLec2 PDFAndrés Camilo LópezNo ratings yet
- Cement Kiln Pyro BalanceDocument44 pagesCement Kiln Pyro BalanceirfanNo ratings yet
- Axle Order Form: Axles Wheel Stud InformationDocument2 pagesAxle Order Form: Axles Wheel Stud InformationHaytham AjibNo ratings yet
- Iso 11464Document15 pagesIso 11464zvjesosNo ratings yet
- DEH Governor - Mitsubishi Heavy Industries, LTD PDFDocument2 pagesDEH Governor - Mitsubishi Heavy Industries, LTD PDFhamidkatebi100% (1)
- Anchor Scan ParametersDocument3 pagesAnchor Scan ParametersDesmon TutuNo ratings yet
- A Description of Dzә (Jenjo) Nouns and Noun PhrasesDocument15 pagesA Description of Dzә (Jenjo) Nouns and Noun PhrasesSamuel EkpoNo ratings yet
- Rise of Health and Medical Tourism by John ConnellDocument19 pagesRise of Health and Medical Tourism by John ConnellEren Osman KurnazNo ratings yet
- Edited - GROUP GAMES - COURSE SYLLABUS 1st Sem AY2023 2024Document7 pagesEdited - GROUP GAMES - COURSE SYLLABUS 1st Sem AY2023 2024B09 Kurt Benedict HillNo ratings yet
- Lesson Plan Template - Social EmotionalDocument2 pagesLesson Plan Template - Social Emotionalapi-573540610No ratings yet
- Motion in Space: Velocity and AccelerationDocument34 pagesMotion in Space: Velocity and AccelerationCrystal MaxNo ratings yet
- E-Proceedings - ICCRET-2023Document30 pagesE-Proceedings - ICCRET-2023RAGHIB R SHARIFNo ratings yet
- Ardunio Based Home Kitchen Air Monitoring Systm: Presented byDocument25 pagesArdunio Based Home Kitchen Air Monitoring Systm: Presented byManik SharmaNo ratings yet
- Book Order FormDocument2 pagesBook Order Formbinaya BehuraNo ratings yet
- Project 4Document6 pagesProject 4Trai TranNo ratings yet
- Dialogo en Ingles Evidencia 2 Actividad 3Document3 pagesDialogo en Ingles Evidencia 2 Actividad 3CAMILA KEIDY RESTREPO ZAPATANo ratings yet
- Magnetic Electricity GeneratorDocument3 pagesMagnetic Electricity GeneratorShahriar RahmanNo ratings yet
- DanielsDocument5 pagesDanielstv_whiteboyNo ratings yet
- Physics - XIIDocument3 pagesPhysics - XIIPrabith GuptaNo ratings yet
- Mechanical Systems and Signal ProcessingDocument18 pagesMechanical Systems and Signal ProcessingsamueleNo ratings yet
- Bahir Dar Chapter FourDocument54 pagesBahir Dar Chapter Fourblackhat0917No ratings yet
- Mgtowsolution PDFDocument78 pagesMgtowsolution PDFsmit suman100% (2)
- Peter GärdenforsDocument9 pagesPeter GärdenforsusamaknightNo ratings yet
- Kisssoft Tut 016 E WormgearDocument16 pagesKisssoft Tut 016 E WormgearIbraheem KhressNo ratings yet
- Tomb Raider Underworld (Europe) (PC) (En, FR, De, Es, It) (v1.1) - Crystal Dynamics - Free Download, Borrow, ADocument1 pageTomb Raider Underworld (Europe) (PC) (En, FR, De, Es, It) (v1.1) - Crystal Dynamics - Free Download, Borrow, Afrancisco perezNo ratings yet
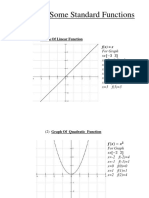
- Lecture 2 Graphs of Some Standard Functions, Shifting of GraphsDocument12 pagesLecture 2 Graphs of Some Standard Functions, Shifting of Graphsghazi membersNo ratings yet
- Procedures For Project Formulation and Management (PPFM) in DrdoDocument86 pagesProcedures For Project Formulation and Management (PPFM) in Drdomkarya_247850155No ratings yet
- (Computer Science Project) To Generate Electricity Bill For Computer Science ProjectDocument33 pages(Computer Science Project) To Generate Electricity Bill For Computer Science ProjectHargur BediNo ratings yet
- Z:/windchill/codebase Z:/windchill/codebaseDocument3 pagesZ:/windchill/codebase Z:/windchill/codebaseamalNo ratings yet
- Shilpa PPT FinalDocument51 pagesShilpa PPT FinalDrakeNo ratings yet






























































































Download as pdf or txt
You might also like
- BYUOpticsBook 2013Document344 pagesBYUOpticsBook 2013geopaok165No ratings yet
- Dynamic Response of Fixed Offshore Structures Under Environmental LoadsDocument16 pagesDynamic Response of Fixed Offshore Structures Under Environmental LoadsxautraixxxxxxNo ratings yet
- 01 Main Part1 2Document68 pages01 Main Part1 2kashyapNo ratings yet
- Journal of Alloys and Compounds: ReviewDocument11 pagesJournal of Alloys and Compounds: ReviewTalita SouzaNo ratings yet
- 147-Waves, Modes, CommunicationsDocument147 pages147-Waves, Modes, CommunicationsRuifeng XuNo ratings yet
- 232 EtdDocument92 pages232 Etdjan dildoNo ratings yet
- Research PaperDocument49 pagesResearch Paperben0706No ratings yet
- Ktab Mo3ama9 Fih KolachDocument186 pagesKtab Mo3ama9 Fih KolachMustapha OuldacheNo ratings yet
- Non Imagistic Optics Aop-10-2-4841Document29 pagesNon Imagistic Optics Aop-10-2-4841Danut StanciuNo ratings yet
- Carlos S. Kubrusly (Auth.) - Spectral Theory of Operators On Hilbert Spaces-Birkhäuser Basel (2012) PDFDocument208 pagesCarlos S. Kubrusly (Auth.) - Spectral Theory of Operators On Hilbert Spaces-Birkhäuser Basel (2012) PDFMohamed EL BERKAOUI100% (1)
- Sapt DFT PDFDocument75 pagesSapt DFT PDFAnonymous pa8pSCC15No ratings yet
- Sapt DFT PDFDocument75 pagesSapt DFT PDFAnonymous pa8pSCC15No ratings yet
- BTH2012 OloumiDocument71 pagesBTH2012 OloumiMilton AlvesNo ratings yet
- RMIT University: Metamaterial ResonatorsDocument24 pagesRMIT University: Metamaterial Resonatorsronalme100% (1)
- Said DissertationDocument129 pagesSaid DissertationSaeed AzarNo ratings yet
- Numerical Analysis of A Bisection-Exclusion Method To Find Zeros of Univariate Analytic FunctionsDocument39 pagesNumerical Analysis of A Bisection-Exclusion Method To Find Zeros of Univariate Analytic FunctionsLucas SantosNo ratings yet
- Malhotra Et Al 2016Document17 pagesMalhotra Et Al 2016Abhishek MalhotraNo ratings yet
- MT5 PDFDocument80 pagesMT5 PDFAnonymous 8VIwRGNo ratings yet
- Jiang 2017Document49 pagesJiang 2017Agtc TandayNo ratings yet
- Computational Fluid Dynamics Modelling of Proton Exchange Membrane Fuel CellsDocument29 pagesComputational Fluid Dynamics Modelling of Proton Exchange Membrane Fuel CellsrmvanginkelNo ratings yet
- Fwejk544 9949 FRRDocument93 pagesFwejk544 9949 FRRaicreativeintellectNo ratings yet
- C RauzyDocument162 pagesC Rauzyc_otescuNo ratings yet
- Electromagnetic Metasurfaces: Physics and Applications: Advances in Optics and Photonics June 2019Document101 pagesElectromagnetic Metasurfaces: Physics and Applications: Advances in Optics and Photonics June 2019Jojo NamNo ratings yet
- Electrospinning of Polymer Nanofibers - Effects On Oriented Morphology, Structures and Tensile PropertiesDocument16 pagesElectrospinning of Polymer Nanofibers - Effects On Oriented Morphology, Structures and Tensile PropertiesCamilo A. Suárez BallesterosNo ratings yet
- Investigation of Near-Field Contribution in SBR For Installed Antenna PerformanceDocument80 pagesInvestigation of Near-Field Contribution in SBR For Installed Antenna Performancelaythe_99No ratings yet
- Electric-Magnetic Duality and The Geometric Langlands ProgramDocument231 pagesElectric-Magnetic Duality and The Geometric Langlands ProgramEliacim VelezNo ratings yet
- Welding Methods of PolymerDocument10 pagesWelding Methods of PolymerGOPINATH SNo ratings yet
- 1 s2.0 S1364032113007260 MainDocument19 pages1 s2.0 S1364032113007260 MainkitsomacNo ratings yet
- Dissertation / Doctoral Thesis: Ab-Initio Description of Optical Properties of Semiconductors and NanocrystalsDocument200 pagesDissertation / Doctoral Thesis: Ab-Initio Description of Optical Properties of Semiconductors and NanocrystalsSiabdallahNo ratings yet
- Understanding The Reaction of Nuclear GRDocument25 pagesUnderstanding The Reaction of Nuclear GRSandipGangurdeNo ratings yet
- Spectrochimica Acta Part B: E. Tognoni, G. Cristoforetti, S. Legnaioli, V. PalleschiDocument14 pagesSpectrochimica Acta Part B: E. Tognoni, G. Cristoforetti, S. Legnaioli, V. PalleschibetjodaNo ratings yet
- High-Contrast Gratings For Integrated Optoelectronics: Connie J. Chang-Hasnain and Weijian YangDocument62 pagesHigh-Contrast Gratings For Integrated Optoelectronics: Connie J. Chang-Hasnain and Weijian YangMohit SinhaNo ratings yet
- FerritesobtainedbysolgelDocument42 pagesFerritesobtainedbysolgelTasnuva HumairaNo ratings yet
- Analogue Gravity Living ReviewDocument159 pagesAnalogue Gravity Living ReviewTapas DasNo ratings yet
- 1 s2.0 S0950061818330307 MainDocument15 pages1 s2.0 S0950061818330307 MainAKINMADE OLUWATOSINNo ratings yet
- Production of Carbon Nanostructures in Biochar BioDocument15 pagesProduction of Carbon Nanostructures in Biochar BioJatin Kumar SaxenaNo ratings yet
- Mbithi Nelson Matheka PHD 2022Document88 pagesMbithi Nelson Matheka PHD 2022Jenő MiklayNo ratings yet
- Bi Metallic NPs by Chemical MethodDocument251 pagesBi Metallic NPs by Chemical Methodmuhammad siddiq siddiqNo ratings yet
- Progress in Energy and Combustion Science: L.Y.M. Gicquel, G. Staffelbach, T. PoinsotDocument36 pagesProgress in Energy and Combustion Science: L.Y.M. Gicquel, G. Staffelbach, T. PoinsotMoses DevaprasannaNo ratings yet
- 315 TextDocument144 pages315 TextKevin PascualNo ratings yet
- 2012 ELSEVIER XRD In-Situ TestDocument13 pages2012 ELSEVIER XRD In-Situ Testirem AydoganNo ratings yet
- Full TextDocument190 pagesFull Textbsmalah11alroxnamNo ratings yet
- Study of SILAR Deposited ZnO For InverteDocument61 pagesStudy of SILAR Deposited ZnO For InvertekardesimnaberyaNo ratings yet
- Progress in Energy and Combustion Science: Kehang Cui, Shigeo MaruyamaDocument21 pagesProgress in Energy and Combustion Science: Kehang Cui, Shigeo MaruyamaAli AliNo ratings yet
- Limousin, G Et Al. Sorption Isotherms A Review On Physical Bases, Modeling and MeasurementDocument27 pagesLimousin, G Et Al. Sorption Isotherms A Review On Physical Bases, Modeling and MeasurementbrianNo ratings yet
- PVDF HFP 1Document24 pagesPVDF HFP 1salmanNo ratings yet
- A Comparative Study of Total Direct and Diffuse Solar Irradiance byDocument11 pagesA Comparative Study of Total Direct and Diffuse Solar Irradiance bysasa_22No ratings yet
- Materials Science and Engineering C: ReviewDocument16 pagesMaterials Science and Engineering C: ReviewBeesam Ramesh KumarNo ratings yet
- Quantum Hadrodynamics and Dense Matter ThesisDocument135 pagesQuantum Hadrodynamics and Dense Matter ThesisalexisNo ratings yet
- Collections of Math IIDocument561 pagesCollections of Math IIHenry GarrettNo ratings yet
- Cooper and BistDocument21 pagesCooper and BistDiana PleşcaNo ratings yet
- Scaling Laws and Electron Properties in Hall Effect ThrustersDocument176 pagesScaling Laws and Electron Properties in Hall Effect Thrustersaakash30janNo ratings yet
- Seddon 2017Document74 pagesSeddon 2017Rayhan XhanNo ratings yet
- Trig BookDocument203 pagesTrig BookmayNo ratings yet
- DEREK R.MILLER - Nanoscale Metal Oxide-Based Heterojunctions For Gas Sensing - Review PDFDocument23 pagesDEREK R.MILLER - Nanoscale Metal Oxide-Based Heterojunctions For Gas Sensing - Review PDFAna Maria NiculescuNo ratings yet
- Journal of Materiomics: Michael Green, Xiaobo ChenDocument39 pagesJournal of Materiomics: Michael Green, Xiaobo ChenSohail AhmedNo ratings yet
- Silicon 2Document221 pagesSilicon 2cafierroNo ratings yet
- Organic and Perovskite Solar Cells Working Principles, Materials and Interfaces - Marinova - 2016Document17 pagesOrganic and Perovskite Solar Cells Working Principles, Materials and Interfaces - Marinova - 2016Aniello LangellaNo ratings yet
- ICTAC Kinetics Committee Recommendations For Performing Kinetic Computations On Thermal Analysis DataDocument19 pagesICTAC Kinetics Committee Recommendations For Performing Kinetic Computations On Thermal Analysis Datatotenkopf0424No ratings yet
- The Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionFrom EverandThe Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionNo ratings yet
- The Action Principle and Partial Differential Equations. (AM-146), Volume 146From EverandThe Action Principle and Partial Differential Equations. (AM-146), Volume 146No ratings yet
- Patil2012 PDFDocument4 pagesPatil2012 PDFAndrés Camilo LópezNo ratings yet
- Study On The Concrete in Chloride Environment Based On Electrochemical Impedance SpectrosDocument13 pagesStudy On The Concrete in Chloride Environment Based On Electrochemical Impedance SpectrosAndrés Camilo LópezNo ratings yet
- Review ArticleDocument20 pagesReview ArticleAndrés Camilo LópezNo ratings yet
- Pu 2005Document6 pagesPu 2005Andrés Camilo LópezNo ratings yet
- Latex Template For Preparing A Research Article For Submission To The Journal of Optical Communications and NetworkingDocument3 pagesLatex Template For Preparing A Research Article For Submission To The Journal of Optical Communications and NetworkingAndrés Camilo LópezNo ratings yet
- Global Hybrid Functionals: A Look at The Engine Under The HoodDocument16 pagesGlobal Hybrid Functionals: A Look at The Engine Under The HoodAndrés Camilo LópezNo ratings yet
- Does A Photochemical Reaction Have A Reaction Order?: S. R. LoganDocument1 pageDoes A Photochemical Reaction Have A Reaction Order?: S. R. LoganAndrés Camilo LópezNo ratings yet
- Lec2 PDFDocument4 pagesLec2 PDFAndrés Camilo LópezNo ratings yet
- Cement Kiln Pyro BalanceDocument44 pagesCement Kiln Pyro BalanceirfanNo ratings yet
- Axle Order Form: Axles Wheel Stud InformationDocument2 pagesAxle Order Form: Axles Wheel Stud InformationHaytham AjibNo ratings yet
- Iso 11464Document15 pagesIso 11464zvjesosNo ratings yet
- DEH Governor - Mitsubishi Heavy Industries, LTD PDFDocument2 pagesDEH Governor - Mitsubishi Heavy Industries, LTD PDFhamidkatebi100% (1)
- Anchor Scan ParametersDocument3 pagesAnchor Scan ParametersDesmon TutuNo ratings yet
- A Description of Dzә (Jenjo) Nouns and Noun PhrasesDocument15 pagesA Description of Dzә (Jenjo) Nouns and Noun PhrasesSamuel EkpoNo ratings yet
- Rise of Health and Medical Tourism by John ConnellDocument19 pagesRise of Health and Medical Tourism by John ConnellEren Osman KurnazNo ratings yet
- Edited - GROUP GAMES - COURSE SYLLABUS 1st Sem AY2023 2024Document7 pagesEdited - GROUP GAMES - COURSE SYLLABUS 1st Sem AY2023 2024B09 Kurt Benedict HillNo ratings yet
- Lesson Plan Template - Social EmotionalDocument2 pagesLesson Plan Template - Social Emotionalapi-573540610No ratings yet
- Motion in Space: Velocity and AccelerationDocument34 pagesMotion in Space: Velocity and AccelerationCrystal MaxNo ratings yet
- E-Proceedings - ICCRET-2023Document30 pagesE-Proceedings - ICCRET-2023RAGHIB R SHARIFNo ratings yet
- Ardunio Based Home Kitchen Air Monitoring Systm: Presented byDocument25 pagesArdunio Based Home Kitchen Air Monitoring Systm: Presented byManik SharmaNo ratings yet
- Book Order FormDocument2 pagesBook Order Formbinaya BehuraNo ratings yet
- Project 4Document6 pagesProject 4Trai TranNo ratings yet
- Dialogo en Ingles Evidencia 2 Actividad 3Document3 pagesDialogo en Ingles Evidencia 2 Actividad 3CAMILA KEIDY RESTREPO ZAPATANo ratings yet
- Magnetic Electricity GeneratorDocument3 pagesMagnetic Electricity GeneratorShahriar RahmanNo ratings yet
- DanielsDocument5 pagesDanielstv_whiteboyNo ratings yet
- Physics - XIIDocument3 pagesPhysics - XIIPrabith GuptaNo ratings yet
- Mechanical Systems and Signal ProcessingDocument18 pagesMechanical Systems and Signal ProcessingsamueleNo ratings yet
- Bahir Dar Chapter FourDocument54 pagesBahir Dar Chapter Fourblackhat0917No ratings yet
- Mgtowsolution PDFDocument78 pagesMgtowsolution PDFsmit suman100% (2)
- Peter GärdenforsDocument9 pagesPeter GärdenforsusamaknightNo ratings yet
- Kisssoft Tut 016 E WormgearDocument16 pagesKisssoft Tut 016 E WormgearIbraheem KhressNo ratings yet
- Tomb Raider Underworld (Europe) (PC) (En, FR, De, Es, It) (v1.1) - Crystal Dynamics - Free Download, Borrow, ADocument1 pageTomb Raider Underworld (Europe) (PC) (En, FR, De, Es, It) (v1.1) - Crystal Dynamics - Free Download, Borrow, Afrancisco perezNo ratings yet
- Lecture 2 Graphs of Some Standard Functions, Shifting of GraphsDocument12 pagesLecture 2 Graphs of Some Standard Functions, Shifting of Graphsghazi membersNo ratings yet
- Procedures For Project Formulation and Management (PPFM) in DrdoDocument86 pagesProcedures For Project Formulation and Management (PPFM) in Drdomkarya_247850155No ratings yet
- (Computer Science Project) To Generate Electricity Bill For Computer Science ProjectDocument33 pages(Computer Science Project) To Generate Electricity Bill For Computer Science ProjectHargur BediNo ratings yet
- Z:/windchill/codebase Z:/windchill/codebaseDocument3 pagesZ:/windchill/codebase Z:/windchill/codebaseamalNo ratings yet
- Shilpa PPT FinalDocument51 pagesShilpa PPT FinalDrakeNo ratings yet