Download as pdf or txt
You might also like
- Can't Hurt Me by David GogginsDocument5 pagesCan't Hurt Me by David GogginsInfinit1376% (17)
- Articulo#3 2007 (201) Pag 4Document3 pagesArticulo#3 2007 (201) Pag 4Sebastian LeonNo ratings yet
- Whitaker 2003Document5 pagesWhitaker 2003Thati PonceNo ratings yet
- Batten Disease: Features To Facilitate Early Diagnosis: Scientific ReportDocument6 pagesBatten Disease: Features To Facilitate Early Diagnosis: Scientific ReportMaferNo ratings yet
- Etiology of Congenital ScoliosisDocument3 pagesEtiology of Congenital ScoliosisCrissu CrissNo ratings yet
- Treachercollinssyndrome: Albaraa Aljerian,, Mirko S. GilardinoDocument9 pagesTreachercollinssyndrome: Albaraa Aljerian,, Mirko S. GilardinoSai KrupaNo ratings yet
- Cleidocraneal Dysplasia 2000Document3 pagesCleidocraneal Dysplasia 2000Daniela Paz Valenzuela ColmenaresNo ratings yet
- Aaos Pediatric 2020Document57 pagesAaos Pediatric 2020ABUBAKER ZANBOUZINo ratings yet
- 4 Epilepsy in A PatientDocument4 pages4 Epilepsy in A Patientarturschander3614No ratings yet
- Osteopathia Striata With Cranial SclerosisDocument5 pagesOsteopathia Striata With Cranial SclerosisritvikNo ratings yet
- ArticleDocument8 pagesArticleLusian VissNo ratings yet
- A Sonographic Approach To The Prenatal Diagnosis of Skeletal DysplasiasDocument19 pagesA Sonographic Approach To The Prenatal Diagnosis of Skeletal DysplasiasminhhaiNo ratings yet
- CTEV A Review of Current ManagementlDocument6 pagesCTEV A Review of Current Managementlvicky174No ratings yet
- Compile Pediatric ConditionsDocument293 pagesCompile Pediatric ConditionsFatinnadhirah YazimNo ratings yet
- 3I. Scoiuosis: AnomaliesDocument7 pages3I. Scoiuosis: AnomaliesValentina OprisanNo ratings yet
- Legg-Calvé-Perthes DiseaseDocument59 pagesLegg-Calvé-Perthes DiseaseCatalina Elena González CarvajalNo ratings yet
- Brachial Plexus Birth Palsy: An Overview of Early Treatment ConsiderationsDocument7 pagesBrachial Plexus Birth Palsy: An Overview of Early Treatment Considerationsphannghia.ydsNo ratings yet
- Syndromic Craniosynostosis 19pDocument6 pagesSyndromic Craniosynostosis 19pAnna Karolinne NascimentoNo ratings yet
- Spinal Epidural Abscess in Two CalvesDocument8 pagesSpinal Epidural Abscess in Two CalvesRachel AutranNo ratings yet
- S D A o (Iii) : S G D, N T D, D D oDocument10 pagesS D A o (Iii) : S G D, N T D, D D oathayafebNo ratings yet
- Uncommon Disorders Masquerading As Acute Flaccid Paralysis in ChildrenDocument7 pagesUncommon Disorders Masquerading As Acute Flaccid Paralysis in ChildrenAnonymous G20oAbl6p8No ratings yet
- A New Type of Autosomal Recessive Spondyloepiphyseal Dysplasia TardaDocument4 pagesA New Type of Autosomal Recessive Spondyloepiphyseal Dysplasia TardaAldunIdhunNo ratings yet
- Yanko Et Al 2023 Failure of Mandibular Distraction Osteogenesis in Klippel Feil Syndrome 4 A Case Report of A RareDocument6 pagesYanko Et Al 2023 Failure of Mandibular Distraction Osteogenesis in Klippel Feil Syndrome 4 A Case Report of A Rareelizabethhanna045No ratings yet
- Ahmed, Khamis - 2019 - Poland Syndrome A Rare Case Report From PalestineDocument5 pagesAhmed, Khamis - 2019 - Poland Syndrome A Rare Case Report From PalestineshyamNo ratings yet
- Pallister-Killian Mosaic Syndrome in An Omani NewbDocument5 pagesPallister-Killian Mosaic Syndrome in An Omani NewbFrank Harry LampardNo ratings yet
- European Journal of Medical GeneticsDocument6 pagesEuropean Journal of Medical GeneticsLarissa DmtNo ratings yet
- Heterogeneity in Klippel-Feil Syndrome: A New ClassificationDocument8 pagesHeterogeneity in Klippel-Feil Syndrome: A New Classificationjessyramirez07No ratings yet
- Cri Du ChatDocument16 pagesCri Du ChatJet LeeNo ratings yet
- Frontal Lobe SeizuresDocument11 pagesFrontal Lobe Seizuresjhonny churata huarachiNo ratings yet
- Auditory ERPs Reveal Brain Dysfunction in Infants With PlagiocephalyDocument6 pagesAuditory ERPs Reveal Brain Dysfunction in Infants With PlagiocephalychiaraNo ratings yet
- C5 Palsy After Cervical Laminectomy: Natural History in A 10-Year SeriesDocument6 pagesC5 Palsy After Cervical Laminectomy: Natural History in A 10-Year SeriesAhmad SyahmiNo ratings yet
- Vgygcc BHJJJJDocument4 pagesVgygcc BHJJJJVsbshNo ratings yet
- Limited Value of Plain Radiographs in Infant Torticollis: PEDIATRICS January 2007Document9 pagesLimited Value of Plain Radiographs in Infant Torticollis: PEDIATRICS January 2007Juan Pablo PérezNo ratings yet
- RJN 2021 4 Art-18Document4 pagesRJN 2021 4 Art-18Emilia LunguNo ratings yet
- Survey-62-P770 Keratoconus Review - QueratoconoDocument14 pagesSurvey-62-P770 Keratoconus Review - QueratoconoMaría Antonela NicolóNo ratings yet
- Congenitalmyopathies Andmusculardystrophies: Heather R. Gilbreath,, Diana Castro,, Susan T. IannacconeDocument15 pagesCongenitalmyopathies Andmusculardystrophies: Heather R. Gilbreath,, Diana Castro,, Susan T. IannacconeRizki Nandasari SulbahriNo ratings yet
- International Journal of Research in Pharmaceutical SciencesDocument12 pagesInternational Journal of Research in Pharmaceutical SciencesMagfirah AliyyaNo ratings yet
- Case Report FinalDocument14 pagesCase Report FinalAaron Lemuel OngNo ratings yet
- Magnetic Resonance Imaging Findings in Cerebral Palsy: R Yin, Ds Reddihough, MR Ditchfield and KJ CollinsDocument6 pagesMagnetic Resonance Imaging Findings in Cerebral Palsy: R Yin, Ds Reddihough, MR Ditchfield and KJ CollinsKriti ShuklaNo ratings yet
- 226-Article Text-717-3-10-20141213Document5 pages226-Article Text-717-3-10-20141213jasleen.jk92No ratings yet
- Juvenile Cataract in Association With Tuberous Sclerosis ComplexDocument6 pagesJuvenile Cataract in Association With Tuberous Sclerosis ComplexAnonymous argZ1ZNo ratings yet
- An Insight Into - Hemifacial MicrosomiaDocument6 pagesAn Insight Into - Hemifacial MicrosomiaIJAR JOURNALNo ratings yet
- Miller2012-How Are Memories RetrievedDocument10 pagesMiller2012-How Are Memories Retrievediulia andreeaNo ratings yet
- Palpatory Diagnosis of Plagiocephaly: Nicette Sergueef, Kenneth E. Nelson, Thomas GlonekDocument10 pagesPalpatory Diagnosis of Plagiocephaly: Nicette Sergueef, Kenneth E. Nelson, Thomas GlonekDiana SchlittlerNo ratings yet
- Slipped Capital Femoral EpiphysisDocument43 pagesSlipped Capital Femoral EpiphysisFrancisca VillalobosNo ratings yet
- Ocular Features of Marfan Syndrome: Gordana Stanković-Babić, Milena Vujanović, Jasmina Đorđević-Jocić, Sonja CekićDocument4 pagesOcular Features of Marfan Syndrome: Gordana Stanković-Babić, Milena Vujanović, Jasmina Đorđević-Jocić, Sonja CekićmongiiiNo ratings yet
- Acute Brain MRIDocument7 pagesAcute Brain MRIRobin SianiparNo ratings yet
- New Clinical and Therapeutic Perspectives in Currarino Syndrome (Study of 29 Cases)Document6 pagesNew Clinical and Therapeutic Perspectives in Currarino Syndrome (Study of 29 Cases)Luis Enrique Zea SalazarNo ratings yet
- Ossi Fication of The Posterior Longitudinal Ligament: Etiology, Diagnosis, and Outcomes of Nonoperative and Operative ManagementDocument10 pagesOssi Fication of The Posterior Longitudinal Ligament: Etiology, Diagnosis, and Outcomes of Nonoperative and Operative ManagementWahyu Satria KurniawanNo ratings yet
- Neonatal Cataract: Aetiology, Pathogenesis and ManagementDocument13 pagesNeonatal Cataract: Aetiology, Pathogenesis and ManagementLorenaNo ratings yet
- Dev Riend T 1998Document5 pagesDev Riend T 1998Sarly FebrianaNo ratings yet
- Semionov Imaging of Thoracic WallDocument13 pagesSemionov Imaging of Thoracic Walllaila.forestaNo ratings yet
- Marinesco-Sjo Gren Syndrome - 2013Document5 pagesMarinesco-Sjo Gren Syndrome - 2013Irfan RazaNo ratings yet
- Case Report: A Case of Caudal Regression Syndrome: Walking or Sitting?Document5 pagesCase Report: A Case of Caudal Regression Syndrome: Walking or Sitting?LisnainiMuchlisNo ratings yet
- A Comparison of Fundus Autofluorescence and Retinal Structure in Patients With Stargardt DiseaseDocument7 pagesA Comparison of Fundus Autofluorescence and Retinal Structure in Patients With Stargardt DiseaseEvelyn SepulvedaNo ratings yet
- Cerebral Palsy: Peggy S. Eicher, MO, and Mark Batshaw, MODocument15 pagesCerebral Palsy: Peggy S. Eicher, MO, and Mark Batshaw, MOCamii AlbornozNo ratings yet
- Cervical AuricleDocument2 pagesCervical AuricleTahir S.MNo ratings yet
- Confirmation of The Catania Brachydactylous Type of Acrofacial DysostosisDocument4 pagesConfirmation of The Catania Brachydactylous Type of Acrofacial DysostosisSergioFernandezNo ratings yet
- SWAIMANDocument18 pagesSWAIMANVan John MagallanesNo ratings yet
- Scoliosis in Patients With Friedreich's Ataxia: SpineDocument6 pagesScoliosis in Patients With Friedreich's Ataxia: SpineFamilia Macovei Haralambie-AndreeaNo ratings yet
- A Simple Guide to Klippel-Feil Syndrome, Diagnosis, Treatment and Related ConditionsFrom EverandA Simple Guide to Klippel-Feil Syndrome, Diagnosis, Treatment and Related ConditionsNo ratings yet
- WASHBURNDocument8 pagesWASHBURNalanna07No ratings yet
- "A Vehicle of Symbols and Nothing More" George Romanes, Theory of Mind, Information, and Samuel ButlerDocument31 pages"A Vehicle of Symbols and Nothing More" George Romanes, Theory of Mind, Information, and Samuel Butleralanna07No ratings yet
- Revista Latinoamericana de Psicología 0120-0534: Issn: Direccion - Rlp@konradlorenz - Edu.coDocument18 pagesRevista Latinoamericana de Psicología 0120-0534: Issn: Direccion - Rlp@konradlorenz - Edu.coalanna07No ratings yet
- Le in Inger 1988Document9 pagesLe in Inger 1988alanna07No ratings yet
- Craig 2011Document9 pagesCraig 2011alanna07No ratings yet
- Sullivan 2005Document21 pagesSullivan 2005alanna07No ratings yet
- Gallup JR 1977Document11 pagesGallup JR 1977alanna07No ratings yet
- Does Climate or Mobility Explain The Differences in Body Proportions Between Neandertals and Their Upper Paleolithic Successors?Document6 pagesDoes Climate or Mobility Explain The Differences in Body Proportions Between Neandertals and Their Upper Paleolithic Successors?alanna07No ratings yet
- Genes, Culture, and Agriculture - An Example of Human Niche ConstructionDocument38 pagesGenes, Culture, and Agriculture - An Example of Human Niche Constructionalanna07No ratings yet
- The Amazing Case of Phineas Gage: Projected The Tamping RodDocument2 pagesThe Amazing Case of Phineas Gage: Projected The Tamping Rodalanna07No ratings yet
- SDM-v1 0Document32 pagesSDM-v1 0Franz NussmannNo ratings yet
- Hotel Majestic Kuala Lumpur Press ReleaseDocument3 pagesHotel Majestic Kuala Lumpur Press Releasesam07rocksNo ratings yet
- G6RNDocument3 pagesG6RNValeri Luht Eurocargo FinlandNo ratings yet
- Coal - WikipediaDocument1 pageCoal - WikipediaZalanda NisarNo ratings yet
- Cheadle Area Committee 15th March 2016Document176 pagesCheadle Area Committee 15th March 2016IainRobertsNo ratings yet
- Gate 2000 CyDocument9 pagesGate 2000 CyYocobSamandrewsNo ratings yet
- Psychosexual Theory - Sigmund Freud: Neurologist PsychoanalysisDocument4 pagesPsychosexual Theory - Sigmund Freud: Neurologist PsychoanalysisEmmajean'Ricky GustiloNo ratings yet
- Epaper 20 October 2022Document22 pagesEpaper 20 October 2022Tauya DauramanziNo ratings yet
- Beer & Wine Osd PresDocument18 pagesBeer & Wine Osd PresAnand100% (1)
- Master Circular 16 Compassionate GroundDocument13 pagesMaster Circular 16 Compassionate GroundSHANMUGA VADIVEL SUNDARARAJANNo ratings yet
- OTIS Philadelphia Transit Plan SummaryDocument14 pagesOTIS Philadelphia Transit Plan SummaryKennedy RoseNo ratings yet
- Timber GrowthDocument17 pagesTimber GrowthAbdul Sukur Kamsir100% (3)
- Learning & Teaching MethodologyDocument17 pagesLearning & Teaching MethodologyVaidya Gautham M33% (6)
- Naphthenic Acid / Naphthenate Stabilized Emulsions and The Influence of Crude Oil ComponentsDocument1 pageNaphthenic Acid / Naphthenate Stabilized Emulsions and The Influence of Crude Oil ComponentsLuis Alberto ChirinosNo ratings yet
- Difference Between Dyes and PigmentsDocument5 pagesDifference Between Dyes and PigmentsHameedullah Ansari100% (1)
- Mushroom BoardsDocument4 pagesMushroom BoardsRemyaNo ratings yet
- Edl What Is A TestDocument18 pagesEdl What Is A TestEric Tipton100% (1)
- Presentation 2Document5 pagesPresentation 2TechLakeNo ratings yet
- Busineplan 4Document13 pagesBusineplan 4Irish Gamboa DuenasNo ratings yet
- PCMP (Guzman, Renz N. Bped Ep21)Document4 pagesPCMP (Guzman, Renz N. Bped Ep21)Renz N. GuzmanNo ratings yet
- Combined Cellulitis - FinalDocument78 pagesCombined Cellulitis - Finalsaru_patel0% (1)
- Switchgear CBM ActivitiesDocument62 pagesSwitchgear CBM ActivitiesHelmyNo ratings yet
- Spot LightDocument216 pagesSpot LightCristopherZartaNo ratings yet
- 22-23 Constructive Destructive Study GuideDocument5 pages22-23 Constructive Destructive Study Guideapi-234287636No ratings yet
- 1A Innledning - EPS - Flyer - 2 - Jan - VaslestadDocument4 pages1A Innledning - EPS - Flyer - 2 - Jan - VaslestadNCS40 Trương Quốc BảoNo ratings yet
- Septic Tank Leaching Chamber PDFDocument7 pagesSeptic Tank Leaching Chamber PDFJohan Shane CarsidoNo ratings yet
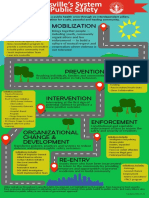
- Reimagining Public SafetyDocument1 pageReimagining Public SafetyWAVE 3 NewsNo ratings yet
- Electrical Inspectorate (11-15)Document5 pagesElectrical Inspectorate (11-15)Aravind NkNo ratings yet
- 2.0 17-02-IM-f (Installation Manual)Document90 pages2.0 17-02-IM-f (Installation Manual)ARMANDOROSAS100% (1)