Download as pdf or txt
You might also like
- Ab Initio and Semiempirical MethodsDocument58 pagesAb Initio and Semiempirical MethodsElisha Niña75% (4)
- OTECDocument6 pagesOTECibong tiriritNo ratings yet
- David Q. Andrews Et Al - Single Molecule Electron Transport Junctions: Charging and Geometric Effects On ConductanceDocument9 pagesDavid Q. Andrews Et Al - Single Molecule Electron Transport Junctions: Charging and Geometric Effects On ConductanceKomodoDSNo ratings yet
- Who, Jan 2011Document5 pagesWho, Jan 2011emediageNo ratings yet
- Heat, Sep 2011Document6 pagesHeat, Sep 2011emediageNo ratings yet
- JAIEE - Volume 8 - Issue 32 - Pages 15-19Document5 pagesJAIEE - Volume 8 - Issue 32 - Pages 15-19majid ghand chiNo ratings yet
- Y. Y. Liang Et Al - Ab Initio Study of Single-Molecule Rotation Switch Based On Nonequilibrium Green's Function TheoryDocument6 pagesY. Y. Liang Et Al - Ab Initio Study of Single-Molecule Rotation Switch Based On Nonequilibrium Green's Function TheoryElectro_LiteNo ratings yet
- Revital Cohen Et Al - Charge Transport in Conjugated Aromatic Molecular Junctions: Molecular Conjugation and Molecule-Electrode CouplingDocument10 pagesRevital Cohen Et Al - Charge Transport in Conjugated Aromatic Molecular Junctions: Molecular Conjugation and Molecule-Electrode CouplingHumdsNo ratings yet
- Enhancement of Thermoelectric Properties of Porphyrin-Based 2021Document7 pagesEnhancement of Thermoelectric Properties of Porphyrin-Based 2021Toshar GargNo ratings yet
- H. Chen Et Al - Control of Substituent Ligand Over Current Through Molecular Devices: An Ab Initio Molecular Orbital TheoryDocument4 pagesH. Chen Et Al - Control of Substituent Ligand Over Current Through Molecular Devices: An Ab Initio Molecular Orbital TheoryElectro_LiteNo ratings yet
- Martin McCullagh Et Al - DNA-Based Optomechanical Molecular MotorDocument8 pagesMartin McCullagh Et Al - DNA-Based Optomechanical Molecular MotorGomsajNo ratings yet
- Chun Zhang Et Al - Current-Voltage Characteristics Through A Single Light-Sensitive MoleculeDocument5 pagesChun Zhang Et Al - Current-Voltage Characteristics Through A Single Light-Sensitive MoleculeGomsajNo ratings yet
- 2019 - Nat. Comm - Impact of Molecular QuadrupoleDocument9 pages2019 - Nat. Comm - Impact of Molecular QuadrupoleBilal NaveedNo ratings yet
- Tomomi Shimazaki Et Al - A Theoretical Study of Molecular Conduction. III. A Nonequilibrium-GreensfunctionDocument12 pagesTomomi Shimazaki Et Al - A Theoretical Study of Molecular Conduction. III. A Nonequilibrium-GreensfunctionGomsajNo ratings yet
- Roi Baer and Daniel Neuhauser - Theoretical Studies of Molecular Scale Near-Field Electron DynamicsDocument9 pagesRoi Baer and Daniel Neuhauser - Theoretical Studies of Molecular Scale Near-Field Electron DynamicsNedsy8No ratings yet
- Hanning Chen Et Al - Classical Electrodynamics Coupled To Quantum Mechanics For Calculation of Molecular Optical Properties: A RT-TDDFT/FDTD ApproachDocument9 pagesHanning Chen Et Al - Classical Electrodynamics Coupled To Quantum Mechanics For Calculation of Molecular Optical Properties: A RT-TDDFT/FDTD ApproachGomsajNo ratings yet
- ZII CO CO 1, Or: Toward A Systematic Molecular Orbital Theory For Excited StatesDocument15 pagesZII CO CO 1, Or: Toward A Systematic Molecular Orbital Theory For Excited StatesIsmael Vargas RodriguezNo ratings yet
- Storey Sochi 2012Document13 pagesStorey Sochi 2012taha_sochiNo ratings yet
- Look, Jan 2011Document5 pagesLook, Jan 2011emediageNo ratings yet
- Density Functional Theory Calculation of Electronic Circular Dichroism Using London OrbitalsDocument10 pagesDensity Functional Theory Calculation of Electronic Circular Dichroism Using London OrbitalsrsgaleanNo ratings yet
- Ground State Energy of Hydrogen-Like Ions in Quantum PlasmasDocument13 pagesGround State Energy of Hydrogen-Like Ions in Quantum PlasmasJit SarkarNo ratings yet
- Roi Baer Et Al - Ab-Initio Study of The AC Impedance of A Molecular JunctionDocument10 pagesRoi Baer Et Al - Ab-Initio Study of The AC Impedance of A Molecular JunctionKomodoDSNo ratings yet
- Alessandro Troisi and Mark A. Ratner - Inelastic Insights For Molecular Tunneling Pathways: Bypassing The Terminal GroupsDocument7 pagesAlessandro Troisi and Mark A. Ratner - Inelastic Insights For Molecular Tunneling Pathways: Bypassing The Terminal GroupsHumdsNo ratings yet
- Brodholt p1049-1053 97Document5 pagesBrodholt p1049-1053 97bencekeNo ratings yet
- Bonding Energy ModelsDocument5 pagesBonding Energy Modelsn5xj70g1xNo ratings yet
- J. Electrochem. Soc.-2017-Baskin-E3438-47Document10 pagesJ. Electrochem. Soc.-2017-Baskin-E3438-47Geovanny JaenzNo ratings yet
- Novel Non-Equilibrium Modelling of A DC Electric Arc in Argon Baeva2016Document17 pagesNovel Non-Equilibrium Modelling of A DC Electric Arc in Argon Baeva2016ElimyNo ratings yet
- ApplPhysLett 84 2409Document4 pagesApplPhysLett 84 2409KaczuHNo ratings yet
- Stochastic Modulation in Molecular Electronic Transport Junctions: Molecular Dynamics Coupled With Charge Transport CalculationsDocument7 pagesStochastic Modulation in Molecular Electronic Transport Junctions: Molecular Dynamics Coupled With Charge Transport CalculationsKomodoDSNo ratings yet
- Refer en CIA 01Document11 pagesRefer en CIA 01complexoNo ratings yet
- J. Franz Et Al - Polarisation Effects in Low-Energy Positron-Molecule ScatteringDocument6 pagesJ. Franz Et Al - Polarisation Effects in Low-Energy Positron-Molecule Scattering4534567No ratings yet
- Analytic Model of The Cathode Region of A Short Glow Discharge in Light GasesDocument16 pagesAnalytic Model of The Cathode Region of A Short Glow Discharge in Light GasesLeonardoNo ratings yet
- Crystals 11 01109Document8 pagesCrystals 11 01109hamza bakkaliNo ratings yet
- tmp2439 TMPDocument12 pagestmp2439 TMPFrontiersNo ratings yet
- Luo 2002Document5 pagesLuo 2002cadolfocastro.ingNo ratings yet
- Quantum Theory of Post-Collision Interaction in Inner-Shell PhotoionizationDocument12 pagesQuantum Theory of Post-Collision Interaction in Inner-Shell PhotoionizationDeepti GuptaNo ratings yet
- 02 Strong-Coupling 2008Document6 pages02 Strong-Coupling 2008NurSalahuddinNo ratings yet
- Steinbach Et Al-1994-Journal of Computational ChemistryDocument17 pagesSteinbach Et Al-1994-Journal of Computational ChemistryDavidFurmanNo ratings yet
- Sina Yeganeh Et Al - Dynamics of Charge Transfer: Rate Processes Formulated With Nonequilibrium Green's FunctionsDocument5 pagesSina Yeganeh Et Al - Dynamics of Charge Transfer: Rate Processes Formulated With Nonequilibrium Green's FunctionsHumdsNo ratings yet
- Gemma C. Solomon Et Al - Understanding Quantum Interference in Coherent Molecular ConductionDocument8 pagesGemma C. Solomon Et Al - Understanding Quantum Interference in Coherent Molecular ConductionKomodoDSNo ratings yet
- Effects of Valence, Geometry and Electronic Correlations On Transport in Transition Metal Benzene Sandwich MoleculesDocument26 pagesEffects of Valence, Geometry and Electronic Correlations On Transport in Transition Metal Benzene Sandwich MoleculesGustavo GuillénNo ratings yet
- Emission and recombination coefficients for hydrogen with κ-distributed electron energiesDocument3 pagesEmission and recombination coefficients for hydrogen with κ-distributed electron energiesjameswhite4321No ratings yet
- Ligand Effects in Heterogeneous Catalysis and ElectrochemistryDocument5 pagesLigand Effects in Heterogeneous Catalysis and ElectrochemistryLuca BrunoNo ratings yet
- Distribution of Electron Charge Centres: A Picture of Bonding Based On Geometric PhasesDocument12 pagesDistribution of Electron Charge Centres: A Picture of Bonding Based On Geometric PhasesMnadeem AkhtarNo ratings yet
- COMSOL SputteringDocument5 pagesCOMSOL SputteringEmanuel CapraNo ratings yet
- Ester Livshits and Roi Baer - A Well-Tempered Density Functional Theory of Electrons in MoleculesDocument10 pagesEster Livshits and Roi Baer - A Well-Tempered Density Functional Theory of Electrons in MoleculesNedsy8No ratings yet
- Ab Initio Study of The Emissive Charge-Transfer States of Solvated Chromophore-Functionalized SilsesquioxanesDocument4 pagesAb Initio Study of The Emissive Charge-Transfer States of Solvated Chromophore-Functionalized Silsesquioxanes蔡承德No ratings yet
- Theoretical Study of The Chemical Bonding in Ni c2Document8 pagesTheoretical Study of The Chemical Bonding in Ni c2NatanaelNo ratings yet
- Ligaduras FuertesDocument9 pagesLigaduras FuertesjulanoNo ratings yet
- New Expressions For Coupling Coefficient Between ResonatorsDocument8 pagesNew Expressions For Coupling Coefficient Between ResonatorsphithucNo ratings yet
- PerovskiteDocument2 pagesPerovskiteDeathEaterReturn100% (1)
- Energy-Loss Functino of TTF-TCNQDocument9 pagesEnergy-Loss Functino of TTF-TCNQWilliamNo ratings yet
- A Direct Method For Locating Minimum-Energy Crossing Points (Mecps) in Spin-Forbidden Transitions and Nonadiabatic ReactionsDocument10 pagesA Direct Method For Locating Minimum-Energy Crossing Points (Mecps) in Spin-Forbidden Transitions and Nonadiabatic ReactionsHarshilGargNo ratings yet
- Oxygen Transport in Perovskite-Type Solid Oxide Fuel Cell Materials: Insights From Quantum MechanicsDocument9 pagesOxygen Transport in Perovskite-Type Solid Oxide Fuel Cell Materials: Insights From Quantum MechanicsrajanadarajanNo ratings yet
- Frank Starrost Et Al - Density-Functional Theory Modeling of Bulk Magnetism With Spin-Dependent PseudopotentialsDocument12 pagesFrank Starrost Et Al - Density-Functional Theory Modeling of Bulk Magnetism With Spin-Dependent PseudopotentialsYamcsaNo ratings yet
- Enhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryDocument3 pagesEnhanced Reactivity of Nanoenergetic Materials: A First-Principles Molecular Dynamics Study Based On Divide-And-Conquer Density Functional TheoryJuaxmawNo ratings yet
- Ncomms 5774Document8 pagesNcomms 5774Samuel ColtNo ratings yet
- Eickerling 2007Document16 pagesEickerling 2007Dianita ValenciaNo ratings yet
- Density Functional Theory and Solvation ModelDocument37 pagesDensity Functional Theory and Solvation Modelsiska tasyaNo ratings yet
- ThesisDocument8 pagesThesisapi-434089240No ratings yet
- Reviews in Computational ChemistryFrom EverandReviews in Computational ChemistryAbby L. ParrillNo ratings yet
- I.K.Gujral Punjab Technical University, Jalandhar: Jalandhar-Kapurthala Highway, JalandharDocument1 pageI.K.Gujral Punjab Technical University, Jalandhar: Jalandhar-Kapurthala Highway, JalandharvijaykumarlambaNo ratings yet
- S.B. Roll NoDocument5 pagesS.B. Roll NovijaykumarlambaNo ratings yet
- CommonDocument10 pagesCommonvijaykumarlambaNo ratings yet
- Mineral and Power Resource Class 8 Mcqs Questions With AnswersDocument14 pagesMineral and Power Resource Class 8 Mcqs Questions With AnswersvijaykumarlambaNo ratings yet
- Chapter 4 Agriculture With Answers: Griculture Class 8 Extra Questions and Answer Geography Chapter 4 VeryDocument9 pagesChapter 4 Agriculture With Answers: Griculture Class 8 Extra Questions and Answer Geography Chapter 4 VeryvijaykumarlambaNo ratings yet
- Chapter 4 Agriculture With Answers: Griculture Class 8 Extra Questions and Answer Geography Chapter 4 VeryDocument6 pagesChapter 4 Agriculture With Answers: Griculture Class 8 Extra Questions and Answer Geography Chapter 4 Veryvijaykumarlamba100% (1)
- Correct AnswerDocument31 pagesCorrect AnswervijaykumarlambaNo ratings yet
- Class VIII: Chapter 1 (Resource) Multiple Choice QuestionsDocument11 pagesClass VIII: Chapter 1 (Resource) Multiple Choice QuestionsvijaykumarlambaNo ratings yet
- Mineral and Power Resource Class 8 Mcqs Questions With AnswersDocument18 pagesMineral and Power Resource Class 8 Mcqs Questions With AnswersvijaykumarlambaNo ratings yet
- Civil GeneralDocument1 pageCivil GeneralvijaykumarlambaNo ratings yet
- Chapter 4 Agriculture With Answers: Griculture Class 8 Extra Questions and Answer Geography Chapter 4 VeryDocument26 pagesChapter 4 Agriculture With Answers: Griculture Class 8 Extra Questions and Answer Geography Chapter 4 VeryvijaykumarlambaNo ratings yet
- Mention The Different Categories Under Which Law Can Be ClassifiedDocument8 pagesMention The Different Categories Under Which Law Can Be ClassifiedvijaykumarlambaNo ratings yet
- Class VIII: Chapter 1 (Resource) Multiple Choice QuestionsDocument24 pagesClass VIII: Chapter 1 (Resource) Multiple Choice QuestionsvijaykumarlambaNo ratings yet
- Mineral and Power Resource Class 8 Mcqs Questions With AnswersDocument28 pagesMineral and Power Resource Class 8 Mcqs Questions With Answersvijaykumarlamba100% (1)
- C 2Document32 pagesC 2vijaykumarlambaNo ratings yet
- Nguyen-Kuok S. - Theory of Low-Temperature Plasma Physics - 2017Document507 pagesNguyen-Kuok S. - Theory of Low-Temperature Plasma Physics - 2017Vantil100% (1)
- Group One ProjectDocument28 pagesGroup One ProjectleeNo ratings yet
- Pharmasutic (Physical Pharmacy) Assignment 01Document14 pagesPharmasutic (Physical Pharmacy) Assignment 01AAMIR NAWAZNo ratings yet
- Timeline of Atomic TheoryDocument1 pageTimeline of Atomic TheoryHyung AynNo ratings yet
- Rotomac Progressive Cavity Pumps: Installation, Operation and Maintenance Manual 'LDocument14 pagesRotomac Progressive Cavity Pumps: Installation, Operation and Maintenance Manual 'Lkallappa naikNo ratings yet
- RWYL102 Operation Manual (FABRICA)Document10 pagesRWYL102 Operation Manual (FABRICA)bifok96010No ratings yet
- Figure 1. The Psychrometric Chart (Low Temperature)Document8 pagesFigure 1. The Psychrometric Chart (Low Temperature)Daniel RusliNo ratings yet
- Shimadzu FTIR 8400s ManualDocument16 pagesShimadzu FTIR 8400s ManualMohammed HamzahNo ratings yet
- Temperature Dependence and PlatinumDocument3 pagesTemperature Dependence and PlatinumTeh Boon SiangNo ratings yet
- 0625 w09 QP 6Document16 pages0625 w09 QP 6koolroNo ratings yet
- Seoni School Design and Drawings With Calculaiton 03-10-22Document48 pagesSeoni School Design and Drawings With Calculaiton 03-10-22danish javedNo ratings yet
- CBSE Class 10 Science Chapter 13 Magnetic Effects of Electric Current Important Questions 2022-23Document34 pagesCBSE Class 10 Science Chapter 13 Magnetic Effects of Electric Current Important Questions 2022-23Rohan SenapathiNo ratings yet
- Lance Design For Argon Bubbling in Molten MetalDocument12 pagesLance Design For Argon Bubbling in Molten MetalJOAN REYES MIRANDANo ratings yet
- Double Shear Single ShearDocument3 pagesDouble Shear Single ShearJoyce ChepkiruiNo ratings yet
- Kegiatan Kalibrasi Tahun: 2022: Puskesmas GUNUNG KALERDocument2 pagesKegiatan Kalibrasi Tahun: 2022: Puskesmas GUNUNG KALERNova ArdiantoNo ratings yet
- Module 1: Shear Strength of Soil: Engr. Vuangh Erick B. Barrantes, MSCDocument34 pagesModule 1: Shear Strength of Soil: Engr. Vuangh Erick B. Barrantes, MSCKenneth Bryan FontanillasNo ratings yet
- Identifikasi Kegiatan Konservasi Di Sektor IndustriDocument98 pagesIdentifikasi Kegiatan Konservasi Di Sektor IndustriHera WahyuniNo ratings yet
- Folio Sains Ting.3 (Chap 9 & 10 + Questions)Document24 pagesFolio Sains Ting.3 (Chap 9 & 10 + Questions)Mr.D-SIM100% (18)
- OPSS 906 Feb93 ElectrodeDocument13 pagesOPSS 906 Feb93 ElectrodeHamid MansouriNo ratings yet
- An Experimental Study On Lightemitting Concrete: Kavya S, Karthik D, Sivaraja MDocument5 pagesAn Experimental Study On Lightemitting Concrete: Kavya S, Karthik D, Sivaraja MBanNo ratings yet
- Bakliwal Tutorials Winter + Sum. Foundation Btest-6 Physics: Single Correct TypeDocument6 pagesBakliwal Tutorials Winter + Sum. Foundation Btest-6 Physics: Single Correct TypeLalani AakashNo ratings yet
- Dewey Physical Properties of Blast Waves 2016Document35 pagesDewey Physical Properties of Blast Waves 2016NgocTraiNguyenNo ratings yet
- Annex 4Document36 pagesAnnex 4Ann Matinong Dela Cruz100% (1)
- Thesis Alex Lotgering Final PublicDocument197 pagesThesis Alex Lotgering Final PublicNaval2014DNNo ratings yet
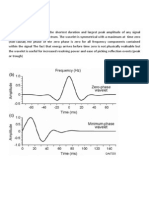
- Zero PhaseDocument4 pagesZero PhaseMuhammad Aftab KhanNo ratings yet
- Heat Exchanger Postlab ReportDocument5 pagesHeat Exchanger Postlab Reportgracebrewster123No ratings yet
- San Roque Dam Static Stability Analysis ReportDocument5 pagesSan Roque Dam Static Stability Analysis ReportIndrian Cyril MendozaNo ratings yet
- Background: Gravity Density VelocityDocument9 pagesBackground: Gravity Density VelocityIrvan Ardyka PrastamaNo ratings yet
- Glass Wool: General InformationDocument1 pageGlass Wool: General InformationYoutube TesterNo ratings yet