
Opening Remarks
Opening Remarks
You might also like
- Clinical Research Vs Clinical PracticeDocument3 pagesClinical Research Vs Clinical PracticeSmita KatheNo ratings yet
- Pharmaceutical and Medical DeviceDocument272 pagesPharmaceutical and Medical Devicemodava100% (1)
- Pan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)Document5 pagesPan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)fdablogNo ratings yet
- Value of Clinical Laboratory Services in Health CareDocument4 pagesValue of Clinical Laboratory Services in Health Careapi-280296463No ratings yet
- Apps To Help You Manage Your Ballya in Vitro Diagnostics Rapid Test ManufacturerDocument3 pagesApps To Help You Manage Your Ballya in Vitro Diagnostics Rapid Test ManufacturerdenopehecpNo ratings yet
- Clinical Trials Its Difficulties and The SolutionDocument8 pagesClinical Trials Its Difficulties and The SolutionSmartway PharmaceuticalsNo ratings yet
- Section Two - Evidence-Based Validation of Safety and EfficacyDocument17 pagesSection Two - Evidence-Based Validation of Safety and EfficacyKomal TalrejaNo ratings yet
- Unraveling The Potential of Real-World Studies (2017)Document4 pagesUnraveling The Potential of Real-World Studies (2017)Arturo Lopez GilNo ratings yet
- Clinical Trials Thesis TopicsDocument4 pagesClinical Trials Thesis Topicsshannonjoyarvada100% (2)
- Association For Molecular PathologyDocument4 pagesAssociation For Molecular PathologyfdablogNo ratings yet
- July 19-20, 2010Document3 pagesJuly 19-20, 2010fdablogNo ratings yet
- CE of Diagnostic Tests Good Practice Recommendations 1709007663Document14 pagesCE of Diagnostic Tests Good Practice Recommendations 1709007663Sandro PaimNo ratings yet
- Good Clinical Practice Guidelines IndiaDocument4 pagesGood Clinical Practice Guidelines IndiaMonica0% (1)
- INTRODUCTIONDocument6 pagesINTRODUCTIONJoyce Madelle OcsoNo ratings yet
- Fit For Purpose Method DevelopmentDocument17 pagesFit For Purpose Method DevelopmentjakekeiNo ratings yet
- CLINICAL TRIALS DDD Mid StartDocument7 pagesCLINICAL TRIALS DDD Mid Startheyyo ggNo ratings yet
- Brief Review On Clinical TrialsDocument23 pagesBrief Review On Clinical TrialssallurajNo ratings yet
- Exposition On The Importance of Quality Research MethodologyDocument5 pagesExposition On The Importance of Quality Research MethodologyYape SokoNo ratings yet
- Clinical TrialsDocument16 pagesClinical TrialsAndrewTiamoNo ratings yet
- Research 101: Sponsored byDocument34 pagesResearch 101: Sponsored byMohammed HammedNo ratings yet
- Literature Review Hierarchy of EvidenceDocument6 pagesLiterature Review Hierarchy of Evidencec5s8r1zc100% (1)
- What Is EBD?Document7 pagesWhat Is EBD?RATHEESH M. S.No ratings yet
- Topic-Evidence Based Nursing Practice And: Best PracticesDocument15 pagesTopic-Evidence Based Nursing Practice And: Best PracticesgemergencycareNo ratings yet
- Phases of Clinical TrialsDocument17 pagesPhases of Clinical Trialselsaveradqp100% (1)
- StepsDocument4 pagesStepsshrikantNo ratings yet
- JCTLM Global ApproachDocument9 pagesJCTLM Global ApproachElena ShcherbakovaNo ratings yet
- Drug Study Designs - FDADocument4 pagesDrug Study Designs - FDANaydu Rey ArriagaNo ratings yet
- FDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274Document2 pagesFDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274fdablogNo ratings yet
- Stat & ResearchDocument276 pagesStat & ResearchSYED ALI HUSSAINNo ratings yet
- Clinical Trials: FDA ApprovalDocument3 pagesClinical Trials: FDA Approvalthamizh555No ratings yet
- What Is Best Practice?: Caroline BadgerDocument12 pagesWhat Is Best Practice?: Caroline BadgerDražen GrobnikNo ratings yet
- DR Raya Ezat Lec 4Document16 pagesDR Raya Ezat Lec 4Djdjjd SiisusNo ratings yet
- 2015 Fda LDT Workshop CapDocument9 pages2015 Fda LDT Workshop CapTadilakshmikiranNo ratings yet
- Guidance For Conducting Clinical Trials Based On Drugs Medical Products Good Clinical PracticeDocument44 pagesGuidance For Conducting Clinical Trials Based On Drugs Medical Products Good Clinical PracticeMostafa SalahNo ratings yet
- Literature Review Is What Level of EvidenceDocument5 pagesLiterature Review Is What Level of Evidencefahynavakel2100% (1)
- EBP Appraising DiagnosticDocument95 pagesEBP Appraising DiagnostictrimayunikaNo ratings yet
- DI - Intelligent Clinical TrialsDocument36 pagesDI - Intelligent Clinical TrialsPKNo ratings yet
- Real-World Evidence: A Better Life Journey For Pharmas, Payers and PatientsDocument5 pagesReal-World Evidence: A Better Life Journey For Pharmas, Payers and PatientsCognizantNo ratings yet
- Preoperative AssessmentDocument9 pagesPreoperative Assessment1234chocoNo ratings yet
- HIV Vaccine Development: Strategies For Preclinical and Clinical InvestigationDocument6 pagesHIV Vaccine Development: Strategies For Preclinical and Clinical InvestigationErika KusumawatiNo ratings yet
- Ophthalmic Pharmaceutical Clinical Trials: Design ConsiderationsDocument20 pagesOphthalmic Pharmaceutical Clinical Trials: Design ConsiderationsNguyen Minh PhuNo ratings yet
- Familly AmericaDocument8 pagesFamilly AmericaOka Iramda SaputraNo ratings yet
- Insta Labs Limited - PPBM - A8Document18 pagesInsta Labs Limited - PPBM - A8shashank_shekhar_74No ratings yet
- Ejifcc2008vol19no2pp095 105Document11 pagesEjifcc2008vol19no2pp095 105Chamara Nishshanka KasturirathnaNo ratings yet
- Evidence Based Practice (EBP) in NursingDocument31 pagesEvidence Based Practice (EBP) in NursingVictoria Castillo Tamayo100% (1)
- Clinical TrialsDocument51 pagesClinical TrialsRajan JattNo ratings yet
- The Drug Development ProcessDocument7 pagesThe Drug Development ProcessSACHIN BHASKAR NARKHEDE100% (1)
- CQI by CommitteeDocument6 pagesCQI by CommitteeJhOy XiNo ratings yet
- Bertholf2017 Chapter LaboratoryStructureAndFunctionDocument23 pagesBertholf2017 Chapter LaboratoryStructureAndFunctionci8084102No ratings yet
- Survey of Critical Value ReportingDocument6 pagesSurvey of Critical Value ReportingccumplidouNo ratings yet
- RE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsDocument3 pagesRE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsfdablogNo ratings yet
- CDRH2011111 CompanionDx Final Guidance 7-24-14 PDFDocument13 pagesCDRH2011111 CompanionDx Final Guidance 7-24-14 PDFstalker1841No ratings yet
- Documenting Critical Values at The Point of Care (2022) - ADLMDocument3 pagesDocumenting Critical Values at The Point of Care (2022) - ADLMLyonTrioréNo ratings yet
- Understanding and Preparing For Clinical Drug Trial AuditsDocument3 pagesUnderstanding and Preparing For Clinical Drug Trial AuditspekksNo ratings yet
- 2ahandout ENG 2020 ElearningDocument5 pages2ahandout ENG 2020 ElearningNupura AjeshNo ratings yet
- Research Paper On Clinical TrialsDocument4 pagesResearch Paper On Clinical Trialsacpjxhznd100% (1)
- Situational Analysis Lague, Inah Krizia O. BSN 2ADocument3 pagesSituational Analysis Lague, Inah Krizia O. BSN 2Ainah krizia lagueNo ratings yet
- Bty459 CaDocument9 pagesBty459 Cavivekanand879355443612No ratings yet
- Drug Development ProcessapolloDocument19 pagesDrug Development Processapollotamara_0021No ratings yet
- Agendia - Dr. Bernhard Sixt, CeoDocument1 pageAgendia - Dr. Bernhard Sixt, CeofdablogNo ratings yet
- Comments On Regulating LDTDocument2 pagesComments On Regulating LDTfdablogNo ratings yet
- FDA/CDHR Public Meeting: Oversight of Laboratory Developed Tests (Direct-to-Consumer Testing)Document9 pagesFDA/CDHR Public Meeting: Oversight of Laboratory Developed Tests (Direct-to-Consumer Testing)fdablogNo ratings yet
- Small Business Developed Innovative Tests: A Proposed Regulatory Exemption ForDocument4 pagesSmall Business Developed Innovative Tests: A Proposed Regulatory Exemption ForfdablogNo ratings yet
- Georgirene D. Vladutiu, PH.DDocument8 pagesGeorgirene D. Vladutiu, PH.DfdablogNo ratings yet
- About Genomic Health, Inc.: Genomic Health Is Committed To Prolonging and Enhancing The Lives of Patients With CancerDocument7 pagesAbout Genomic Health, Inc.: Genomic Health Is Committed To Prolonging and Enhancing The Lives of Patients With CancerfdablogNo ratings yet
- Mary Del Brady Chairman & Ceo, Cvergenx, Inc. Advisor & Founding Ceo, Redpath Integrated Pathology, Inc. Member, Steering Committee, Coalition For 21 Century MedicineDocument4 pagesMary Del Brady Chairman & Ceo, Cvergenx, Inc. Advisor & Founding Ceo, Redpath Integrated Pathology, Inc. Member, Steering Committee, Coalition For 21 Century MedicinefdablogNo ratings yet
- Outreach and Education: Industry Best Practices: Mya ThomaeDocument11 pagesOutreach and Education: Industry Best Practices: Mya ThomaefdablogNo ratings yet
- Chair, Department of Laboratory Medicine and Pathology, Mayo Clinic President and Chief Executive Officer, Mayo Medical LaboratoriesDocument2 pagesChair, Department of Laboratory Medicine and Pathology, Mayo Clinic President and Chief Executive Officer, Mayo Medical LaboratoriesfdablogNo ratings yet
- From: To: Date: SubjectDocument4 pagesFrom: To: Date: SubjectfdablogNo ratings yet
- Fda 2010 N 0274 0060.1Document2 pagesFda 2010 N 0274 0060.1fdablogNo ratings yet
- FDA Public Meeting On Laboratory Developed Tests July 19, 2010Document3 pagesFDA Public Meeting On Laboratory Developed Tests July 19, 2010fdablogNo ratings yet
- Oversight of LDTS: Clinical Laboratory Challenges: Liz Lison, Advocea, LLCDocument9 pagesOversight of LDTS: Clinical Laboratory Challenges: Liz Lison, Advocea, LLCfdablogNo ratings yet
- FDA/CDRH Public Meeting: Oversight of Laboratory Developed Tests - Patient ConsiderationsDocument8 pagesFDA/CDRH Public Meeting: Oversight of Laboratory Developed Tests - Patient ConsiderationsfdablogNo ratings yet
- Georgirene D. Vladutiu, PH.DDocument9 pagesGeorgirene D. Vladutiu, PH.DfdablogNo ratings yet
- Laboratory Developed TestsDocument10 pagesLaboratory Developed TestsfdablogNo ratings yet
- Association For Molecular PathologyDocument4 pagesAssociation For Molecular PathologyfdablogNo ratings yet
- Appropriateness For Ldts Companion DX Assays: Perspectives On To Be Used AsDocument16 pagesAppropriateness For Ldts Companion DX Assays: Perspectives On To Be Used AsfdablogNo ratings yet
- RE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsDocument3 pagesRE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsfdablogNo ratings yet
- Pan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)Document5 pagesPan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)fdablogNo ratings yet
- TH THDocument2 pagesTH THfdablogNo ratings yet
- Fda Regulation: Lab Developed Tests (LDTS) : Ted SnelgroveDocument6 pagesFda Regulation: Lab Developed Tests (LDTS) : Ted SnelgrovefdablogNo ratings yet
- July 19-20, 2010Document3 pagesJuly 19-20, 2010fdablogNo ratings yet
- FDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274Document2 pagesFDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274fdablogNo ratings yet
- (DIGEST) PBOAP vs. DOLE, G.R. No. 202275, July 17, 2018Document2 pages(DIGEST) PBOAP vs. DOLE, G.R. No. 202275, July 17, 2018Harold Q. GardonNo ratings yet
- Regional Development Plan CAR 2017-2022 Chapter 1Document6 pagesRegional Development Plan CAR 2017-2022 Chapter 1Andrew EvangelistaNo ratings yet
- Gillette India LTD MAK - II Project: Group 6 Section BDocument92 pagesGillette India LTD MAK - II Project: Group 6 Section BPrakhar RatheeNo ratings yet
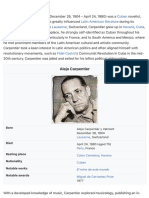
- Alejo Carpentier: Cuban Musicologist Latin American Literature "Boom" Period Lausanne Havana CubaDocument19 pagesAlejo Carpentier: Cuban Musicologist Latin American Literature "Boom" Period Lausanne Havana CubaalexcvsNo ratings yet
- Section 14 HV Network DesignDocument11 pagesSection 14 HV Network DesignUsama AhmedNo ratings yet
- Ethics SyllabusDocument5 pagesEthics SyllabusprincessNo ratings yet
- The Gun Is CivilizationDocument2 pagesThe Gun Is Civilizationandrea-fioreNo ratings yet
- Manual For Conflict AnalysisDocument38 pagesManual For Conflict AnalysisNadine KadriNo ratings yet
- BCA - 17UBC4A4 Computer Based Optimization TechniquesDocument16 pagesBCA - 17UBC4A4 Computer Based Optimization Techniquesabhijeetbhojak1No ratings yet
- Cadila HealthcareDocument3 pagesCadila Healthcarenenu_b2048961No ratings yet
- ¿Por Qué La Tecnología No Puede SalvarnosDocument18 pages¿Por Qué La Tecnología No Puede SalvarnosAle Santizo100% (1)
- Scheme of Work Form 2 English 2018: Week Types Lesson (SOW) Theme Unit (Pulse 2)Document2 pagesScheme of Work Form 2 English 2018: Week Types Lesson (SOW) Theme Unit (Pulse 2)Subramaniam Periannan100% (2)
- capstone-LRRDocument15 pagescapstone-LRRronskierelenteNo ratings yet
- Internship Report of Boss Home AppliancesDocument43 pagesInternship Report of Boss Home ApplianceshhaiderNo ratings yet
- Holy Spirit in Christianity - WikipediaDocument21 pagesHoly Spirit in Christianity - WikipediaSunny BautistaNo ratings yet
- Research Output of Readings in Philippine History 1Document12 pagesResearch Output of Readings in Philippine History 1Itshaaa MeeNo ratings yet
- Forever KnowledgeDocument2 pagesForever KnowledgeminariiNo ratings yet
- The Acient Eurasian World - Vol. IDocument352 pagesThe Acient Eurasian World - Vol. IIpsissimus100% (1)
- Markov Chain PoetryDocument24 pagesMarkov Chain PoetryBea MillwardNo ratings yet
- Soc Psych Research 2Document6 pagesSoc Psych Research 2OSABEL GILLIANNo ratings yet
- Imogene M. King - Theory of Goal AttainmentDocument18 pagesImogene M. King - Theory of Goal AttainmentLouis Gabriel AdayaNo ratings yet
- UntitledDocument18 pagesUntitledjeralyn juditNo ratings yet
- Rizal Life Works BL Guide Quiz 12 and PrelimDocument15 pagesRizal Life Works BL Guide Quiz 12 and PrelimCarla Mae Alcantara100% (1)
- Case Comment On Uttam V Saubhag SinghDocument8 pagesCase Comment On Uttam V Saubhag SinghARJU JAMBHULKARNo ratings yet
- E-Health Care Management Project ReportDocument41 pagesE-Health Care Management Project Reportvikas guptaNo ratings yet
- India Today 09.11.2020Document66 pagesIndia Today 09.11.2020sures108No ratings yet
- Explaining The TrinityDocument3 pagesExplaining The TrinityAriellaNo ratings yet
- Ifma Fmpv3-0 F-B Ch2Document87 pagesIfma Fmpv3-0 F-B Ch2Mohamed IbrahimNo ratings yet
- AC Checklist of ToolsDocument4 pagesAC Checklist of ToolsTvet AcnNo ratings yet
- The Prayer of The Prophet (Sallallaho Alaihe Wa Sallam)Document13 pagesThe Prayer of The Prophet (Sallallaho Alaihe Wa Sallam)Dawah ChannelNo ratings yet












































































































Download as docx, pdf, or txt
You might also like
- Clinical Research Vs Clinical PracticeDocument3 pagesClinical Research Vs Clinical PracticeSmita KatheNo ratings yet
- Pharmaceutical and Medical DeviceDocument272 pagesPharmaceutical and Medical Devicemodava100% (1)
- Pan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)Document5 pagesPan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)fdablogNo ratings yet
- Value of Clinical Laboratory Services in Health CareDocument4 pagesValue of Clinical Laboratory Services in Health Careapi-280296463No ratings yet
- Apps To Help You Manage Your Ballya in Vitro Diagnostics Rapid Test ManufacturerDocument3 pagesApps To Help You Manage Your Ballya in Vitro Diagnostics Rapid Test ManufacturerdenopehecpNo ratings yet
- Clinical Trials Its Difficulties and The SolutionDocument8 pagesClinical Trials Its Difficulties and The SolutionSmartway PharmaceuticalsNo ratings yet
- Section Two - Evidence-Based Validation of Safety and EfficacyDocument17 pagesSection Two - Evidence-Based Validation of Safety and EfficacyKomal TalrejaNo ratings yet
- Unraveling The Potential of Real-World Studies (2017)Document4 pagesUnraveling The Potential of Real-World Studies (2017)Arturo Lopez GilNo ratings yet
- Clinical Trials Thesis TopicsDocument4 pagesClinical Trials Thesis Topicsshannonjoyarvada100% (2)
- Association For Molecular PathologyDocument4 pagesAssociation For Molecular PathologyfdablogNo ratings yet
- July 19-20, 2010Document3 pagesJuly 19-20, 2010fdablogNo ratings yet
- CE of Diagnostic Tests Good Practice Recommendations 1709007663Document14 pagesCE of Diagnostic Tests Good Practice Recommendations 1709007663Sandro PaimNo ratings yet
- Good Clinical Practice Guidelines IndiaDocument4 pagesGood Clinical Practice Guidelines IndiaMonica0% (1)
- INTRODUCTIONDocument6 pagesINTRODUCTIONJoyce Madelle OcsoNo ratings yet
- Fit For Purpose Method DevelopmentDocument17 pagesFit For Purpose Method DevelopmentjakekeiNo ratings yet
- CLINICAL TRIALS DDD Mid StartDocument7 pagesCLINICAL TRIALS DDD Mid Startheyyo ggNo ratings yet
- Brief Review On Clinical TrialsDocument23 pagesBrief Review On Clinical TrialssallurajNo ratings yet
- Exposition On The Importance of Quality Research MethodologyDocument5 pagesExposition On The Importance of Quality Research MethodologyYape SokoNo ratings yet
- Clinical TrialsDocument16 pagesClinical TrialsAndrewTiamoNo ratings yet
- Research 101: Sponsored byDocument34 pagesResearch 101: Sponsored byMohammed HammedNo ratings yet
- Literature Review Hierarchy of EvidenceDocument6 pagesLiterature Review Hierarchy of Evidencec5s8r1zc100% (1)
- What Is EBD?Document7 pagesWhat Is EBD?RATHEESH M. S.No ratings yet
- Topic-Evidence Based Nursing Practice And: Best PracticesDocument15 pagesTopic-Evidence Based Nursing Practice And: Best PracticesgemergencycareNo ratings yet
- Phases of Clinical TrialsDocument17 pagesPhases of Clinical Trialselsaveradqp100% (1)
- StepsDocument4 pagesStepsshrikantNo ratings yet
- JCTLM Global ApproachDocument9 pagesJCTLM Global ApproachElena ShcherbakovaNo ratings yet
- Drug Study Designs - FDADocument4 pagesDrug Study Designs - FDANaydu Rey ArriagaNo ratings yet
- FDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274Document2 pagesFDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274fdablogNo ratings yet
- Stat & ResearchDocument276 pagesStat & ResearchSYED ALI HUSSAINNo ratings yet
- Clinical Trials: FDA ApprovalDocument3 pagesClinical Trials: FDA Approvalthamizh555No ratings yet
- What Is Best Practice?: Caroline BadgerDocument12 pagesWhat Is Best Practice?: Caroline BadgerDražen GrobnikNo ratings yet
- DR Raya Ezat Lec 4Document16 pagesDR Raya Ezat Lec 4Djdjjd SiisusNo ratings yet
- 2015 Fda LDT Workshop CapDocument9 pages2015 Fda LDT Workshop CapTadilakshmikiranNo ratings yet
- Guidance For Conducting Clinical Trials Based On Drugs Medical Products Good Clinical PracticeDocument44 pagesGuidance For Conducting Clinical Trials Based On Drugs Medical Products Good Clinical PracticeMostafa SalahNo ratings yet
- Literature Review Is What Level of EvidenceDocument5 pagesLiterature Review Is What Level of Evidencefahynavakel2100% (1)
- EBP Appraising DiagnosticDocument95 pagesEBP Appraising DiagnostictrimayunikaNo ratings yet
- DI - Intelligent Clinical TrialsDocument36 pagesDI - Intelligent Clinical TrialsPKNo ratings yet
- Real-World Evidence: A Better Life Journey For Pharmas, Payers and PatientsDocument5 pagesReal-World Evidence: A Better Life Journey For Pharmas, Payers and PatientsCognizantNo ratings yet
- Preoperative AssessmentDocument9 pagesPreoperative Assessment1234chocoNo ratings yet
- HIV Vaccine Development: Strategies For Preclinical and Clinical InvestigationDocument6 pagesHIV Vaccine Development: Strategies For Preclinical and Clinical InvestigationErika KusumawatiNo ratings yet
- Ophthalmic Pharmaceutical Clinical Trials: Design ConsiderationsDocument20 pagesOphthalmic Pharmaceutical Clinical Trials: Design ConsiderationsNguyen Minh PhuNo ratings yet
- Familly AmericaDocument8 pagesFamilly AmericaOka Iramda SaputraNo ratings yet
- Insta Labs Limited - PPBM - A8Document18 pagesInsta Labs Limited - PPBM - A8shashank_shekhar_74No ratings yet
- Ejifcc2008vol19no2pp095 105Document11 pagesEjifcc2008vol19no2pp095 105Chamara Nishshanka KasturirathnaNo ratings yet
- Evidence Based Practice (EBP) in NursingDocument31 pagesEvidence Based Practice (EBP) in NursingVictoria Castillo Tamayo100% (1)
- Clinical TrialsDocument51 pagesClinical TrialsRajan JattNo ratings yet
- The Drug Development ProcessDocument7 pagesThe Drug Development ProcessSACHIN BHASKAR NARKHEDE100% (1)
- CQI by CommitteeDocument6 pagesCQI by CommitteeJhOy XiNo ratings yet
- Bertholf2017 Chapter LaboratoryStructureAndFunctionDocument23 pagesBertholf2017 Chapter LaboratoryStructureAndFunctionci8084102No ratings yet
- Survey of Critical Value ReportingDocument6 pagesSurvey of Critical Value ReportingccumplidouNo ratings yet
- RE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsDocument3 pagesRE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsfdablogNo ratings yet
- CDRH2011111 CompanionDx Final Guidance 7-24-14 PDFDocument13 pagesCDRH2011111 CompanionDx Final Guidance 7-24-14 PDFstalker1841No ratings yet
- Documenting Critical Values at The Point of Care (2022) - ADLMDocument3 pagesDocumenting Critical Values at The Point of Care (2022) - ADLMLyonTrioréNo ratings yet
- Understanding and Preparing For Clinical Drug Trial AuditsDocument3 pagesUnderstanding and Preparing For Clinical Drug Trial AuditspekksNo ratings yet
- 2ahandout ENG 2020 ElearningDocument5 pages2ahandout ENG 2020 ElearningNupura AjeshNo ratings yet
- Research Paper On Clinical TrialsDocument4 pagesResearch Paper On Clinical Trialsacpjxhznd100% (1)
- Situational Analysis Lague, Inah Krizia O. BSN 2ADocument3 pagesSituational Analysis Lague, Inah Krizia O. BSN 2Ainah krizia lagueNo ratings yet
- Bty459 CaDocument9 pagesBty459 Cavivekanand879355443612No ratings yet
- Drug Development ProcessapolloDocument19 pagesDrug Development Processapollotamara_0021No ratings yet
- Agendia - Dr. Bernhard Sixt, CeoDocument1 pageAgendia - Dr. Bernhard Sixt, CeofdablogNo ratings yet
- Comments On Regulating LDTDocument2 pagesComments On Regulating LDTfdablogNo ratings yet
- FDA/CDHR Public Meeting: Oversight of Laboratory Developed Tests (Direct-to-Consumer Testing)Document9 pagesFDA/CDHR Public Meeting: Oversight of Laboratory Developed Tests (Direct-to-Consumer Testing)fdablogNo ratings yet
- Small Business Developed Innovative Tests: A Proposed Regulatory Exemption ForDocument4 pagesSmall Business Developed Innovative Tests: A Proposed Regulatory Exemption ForfdablogNo ratings yet
- Georgirene D. Vladutiu, PH.DDocument8 pagesGeorgirene D. Vladutiu, PH.DfdablogNo ratings yet
- About Genomic Health, Inc.: Genomic Health Is Committed To Prolonging and Enhancing The Lives of Patients With CancerDocument7 pagesAbout Genomic Health, Inc.: Genomic Health Is Committed To Prolonging and Enhancing The Lives of Patients With CancerfdablogNo ratings yet
- Mary Del Brady Chairman & Ceo, Cvergenx, Inc. Advisor & Founding Ceo, Redpath Integrated Pathology, Inc. Member, Steering Committee, Coalition For 21 Century MedicineDocument4 pagesMary Del Brady Chairman & Ceo, Cvergenx, Inc. Advisor & Founding Ceo, Redpath Integrated Pathology, Inc. Member, Steering Committee, Coalition For 21 Century MedicinefdablogNo ratings yet
- Outreach and Education: Industry Best Practices: Mya ThomaeDocument11 pagesOutreach and Education: Industry Best Practices: Mya ThomaefdablogNo ratings yet
- Chair, Department of Laboratory Medicine and Pathology, Mayo Clinic President and Chief Executive Officer, Mayo Medical LaboratoriesDocument2 pagesChair, Department of Laboratory Medicine and Pathology, Mayo Clinic President and Chief Executive Officer, Mayo Medical LaboratoriesfdablogNo ratings yet
- From: To: Date: SubjectDocument4 pagesFrom: To: Date: SubjectfdablogNo ratings yet
- Fda 2010 N 0274 0060.1Document2 pagesFda 2010 N 0274 0060.1fdablogNo ratings yet
- FDA Public Meeting On Laboratory Developed Tests July 19, 2010Document3 pagesFDA Public Meeting On Laboratory Developed Tests July 19, 2010fdablogNo ratings yet
- Oversight of LDTS: Clinical Laboratory Challenges: Liz Lison, Advocea, LLCDocument9 pagesOversight of LDTS: Clinical Laboratory Challenges: Liz Lison, Advocea, LLCfdablogNo ratings yet
- FDA/CDRH Public Meeting: Oversight of Laboratory Developed Tests - Patient ConsiderationsDocument8 pagesFDA/CDRH Public Meeting: Oversight of Laboratory Developed Tests - Patient ConsiderationsfdablogNo ratings yet
- Georgirene D. Vladutiu, PH.DDocument9 pagesGeorgirene D. Vladutiu, PH.DfdablogNo ratings yet
- Laboratory Developed TestsDocument10 pagesLaboratory Developed TestsfdablogNo ratings yet
- Association For Molecular PathologyDocument4 pagesAssociation For Molecular PathologyfdablogNo ratings yet
- Appropriateness For Ldts Companion DX Assays: Perspectives On To Be Used AsDocument16 pagesAppropriateness For Ldts Companion DX Assays: Perspectives On To Be Used AsfdablogNo ratings yet
- RE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsDocument3 pagesRE: Docket No. FDA-2010-N-0274 - FDA/CDRH Oversight of Laboratory Developed Tests - Additional CommentsfdablogNo ratings yet
- Pan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)Document5 pagesPan American Society For Clinical Virology Position Statement On Fda Oversight of Laboratory-Developed Tests (LDTS)fdablogNo ratings yet
- TH THDocument2 pagesTH THfdablogNo ratings yet
- Fda Regulation: Lab Developed Tests (LDTS) : Ted SnelgroveDocument6 pagesFda Regulation: Lab Developed Tests (LDTS) : Ted SnelgrovefdablogNo ratings yet
- July 19-20, 2010Document3 pagesJuly 19-20, 2010fdablogNo ratings yet
- FDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274Document2 pagesFDA Oversight of Laboratory Developed Tests Docket Number FDA-2010-N-0274fdablogNo ratings yet
- (DIGEST) PBOAP vs. DOLE, G.R. No. 202275, July 17, 2018Document2 pages(DIGEST) PBOAP vs. DOLE, G.R. No. 202275, July 17, 2018Harold Q. GardonNo ratings yet
- Regional Development Plan CAR 2017-2022 Chapter 1Document6 pagesRegional Development Plan CAR 2017-2022 Chapter 1Andrew EvangelistaNo ratings yet
- Gillette India LTD MAK - II Project: Group 6 Section BDocument92 pagesGillette India LTD MAK - II Project: Group 6 Section BPrakhar RatheeNo ratings yet
- Alejo Carpentier: Cuban Musicologist Latin American Literature "Boom" Period Lausanne Havana CubaDocument19 pagesAlejo Carpentier: Cuban Musicologist Latin American Literature "Boom" Period Lausanne Havana CubaalexcvsNo ratings yet
- Section 14 HV Network DesignDocument11 pagesSection 14 HV Network DesignUsama AhmedNo ratings yet
- Ethics SyllabusDocument5 pagesEthics SyllabusprincessNo ratings yet
- The Gun Is CivilizationDocument2 pagesThe Gun Is Civilizationandrea-fioreNo ratings yet
- Manual For Conflict AnalysisDocument38 pagesManual For Conflict AnalysisNadine KadriNo ratings yet
- BCA - 17UBC4A4 Computer Based Optimization TechniquesDocument16 pagesBCA - 17UBC4A4 Computer Based Optimization Techniquesabhijeetbhojak1No ratings yet
- Cadila HealthcareDocument3 pagesCadila Healthcarenenu_b2048961No ratings yet
- ¿Por Qué La Tecnología No Puede SalvarnosDocument18 pages¿Por Qué La Tecnología No Puede SalvarnosAle Santizo100% (1)
- Scheme of Work Form 2 English 2018: Week Types Lesson (SOW) Theme Unit (Pulse 2)Document2 pagesScheme of Work Form 2 English 2018: Week Types Lesson (SOW) Theme Unit (Pulse 2)Subramaniam Periannan100% (2)
- capstone-LRRDocument15 pagescapstone-LRRronskierelenteNo ratings yet
- Internship Report of Boss Home AppliancesDocument43 pagesInternship Report of Boss Home ApplianceshhaiderNo ratings yet
- Holy Spirit in Christianity - WikipediaDocument21 pagesHoly Spirit in Christianity - WikipediaSunny BautistaNo ratings yet
- Research Output of Readings in Philippine History 1Document12 pagesResearch Output of Readings in Philippine History 1Itshaaa MeeNo ratings yet
- Forever KnowledgeDocument2 pagesForever KnowledgeminariiNo ratings yet
- The Acient Eurasian World - Vol. IDocument352 pagesThe Acient Eurasian World - Vol. IIpsissimus100% (1)
- Markov Chain PoetryDocument24 pagesMarkov Chain PoetryBea MillwardNo ratings yet
- Soc Psych Research 2Document6 pagesSoc Psych Research 2OSABEL GILLIANNo ratings yet
- Imogene M. King - Theory of Goal AttainmentDocument18 pagesImogene M. King - Theory of Goal AttainmentLouis Gabriel AdayaNo ratings yet
- UntitledDocument18 pagesUntitledjeralyn juditNo ratings yet
- Rizal Life Works BL Guide Quiz 12 and PrelimDocument15 pagesRizal Life Works BL Guide Quiz 12 and PrelimCarla Mae Alcantara100% (1)
- Case Comment On Uttam V Saubhag SinghDocument8 pagesCase Comment On Uttam V Saubhag SinghARJU JAMBHULKARNo ratings yet
- E-Health Care Management Project ReportDocument41 pagesE-Health Care Management Project Reportvikas guptaNo ratings yet
- India Today 09.11.2020Document66 pagesIndia Today 09.11.2020sures108No ratings yet
- Explaining The TrinityDocument3 pagesExplaining The TrinityAriellaNo ratings yet
- Ifma Fmpv3-0 F-B Ch2Document87 pagesIfma Fmpv3-0 F-B Ch2Mohamed IbrahimNo ratings yet
- AC Checklist of ToolsDocument4 pagesAC Checklist of ToolsTvet AcnNo ratings yet
- The Prayer of The Prophet (Sallallaho Alaihe Wa Sallam)Document13 pagesThe Prayer of The Prophet (Sallallaho Alaihe Wa Sallam)Dawah ChannelNo ratings yet