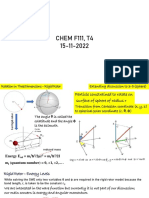
Download as pdf or txt
You might also like
- Pemodelan Inversi Non-Linear Dengan Pendekatan Linear: Pesta Indra Sigalingging, WahyuDocument8 pagesPemodelan Inversi Non-Linear Dengan Pendekatan Linear: Pesta Indra Sigalingging, WahyuPesta SigalinggingNo ratings yet
- Lecture 13Document47 pagesLecture 13--No ratings yet
- Lecture 09Document6 pagesLecture 09bgiangre8372No ratings yet
- Radial and Angular Parts of Atomic OrbitalsDocument7 pagesRadial and Angular Parts of Atomic OrbitalsArchisman MisraNo ratings yet
- 2.01 The Schrodinger Wave Equation For The Hydrogen AtomDocument10 pages2.01 The Schrodinger Wave Equation For The Hydrogen AtomRumbaNo ratings yet
- Chaotic Behaviour of Atomic Energy LevelsDocument7 pagesChaotic Behaviour of Atomic Energy LevelsDele AdigunNo ratings yet
- PHY801HW1SDocument3 pagesPHY801HW1SAnwar Ul HaqNo ratings yet
- Homework 1 - SolutionDocument3 pagesHomework 1 - SolutionIvy JoyceNo ratings yet
- Quantum Mechanics: The Hydrogen Atom: 12th April 2008Document5 pagesQuantum Mechanics: The Hydrogen Atom: 12th April 2008TIMOTHY JAMESONNo ratings yet
- 3.hydrogen Atom Wave FunctionsDocument8 pages3.hydrogen Atom Wave FunctionsSALEEL ENo ratings yet
- Chapter10-Physical Significance of IntegralDocument66 pagesChapter10-Physical Significance of IntegralMika VaughnNo ratings yet
- Wave FN A SDocument5 pagesWave FN A SQwertyNo ratings yet
- 7 The H Ion and Bonding: − ∇ − ∇ − e − e e ψ (r, R) = Eψ (r, R)Document4 pages7 The H Ion and Bonding: − ∇ − ∇ − e − e e ψ (r, R) = Eψ (r, R)alhanunNo ratings yet
- 14-semiconductorsDocument20 pages14-semiconductorssuchit.ijantakarNo ratings yet
- Radial and Angular Parts of Atomic OrbitalsDocument3 pagesRadial and Angular Parts of Atomic OrbitalsW47K3R 27886No ratings yet
- Adobe Scan 20-Dec-2023Document24 pagesAdobe Scan 20-Dec-2023Esha sainiNo ratings yet
- Diatomic Molecule: Vibrational and Rotational SpectraDocument16 pagesDiatomic Molecule: Vibrational and Rotational SpectraCamilo Monsalve MayaNo ratings yet
- BITS Pilani Chemistry Hamilton OperatorDocument11 pagesBITS Pilani Chemistry Hamilton OperatorNaresh SehdevNo ratings yet
- Radial Wave Function and Angular Wave FunctionsDocument8 pagesRadial Wave Function and Angular Wave FunctionsBhavesh Garg100% (2)
- Indian Institute of Technology, Guwahati: CH101 Class 11 Physical ChemistryDocument5 pagesIndian Institute of Technology, Guwahati: CH101 Class 11 Physical ChemistryMihir Kumar MechNo ratings yet
- Quantum Mechanics Course ZeemansplittingDocument29 pagesQuantum Mechanics Course ZeemansplittingjlbalbNo ratings yet
- Atomic Struture - Amit NagDocument4 pagesAtomic Struture - Amit NagKrishna SaxenaNo ratings yet
- Tight-Binding Model of Electronic StructuresDocument9 pagesTight-Binding Model of Electronic StructuresJulian David Henao EscobarNo ratings yet
- Circuits SolnDocument5 pagesCircuits Solndwaytikitan28No ratings yet
- Solutions: Quiz #2 Solutions Page 1 of 6Document6 pagesSolutions: Quiz #2 Solutions Page 1 of 6gorav sharmaNo ratings yet
- Solution To HW ProblemsDocument4 pagesSolution To HW ProblemsAditya PrasadNo ratings yet
- LKya Xko 27 A EQG4 Ei Kyej P2 DCR LM MWKFT9 OHdem YEDocument10 pagesLKya Xko 27 A EQG4 Ei Kyej P2 DCR LM MWKFT9 OHdem YESEBASTIAN CRISTOBAL DOMINGUEZ GALARZANo ratings yet
- Soft Blue KuduDocument10 pagesSoft Blue KuduAditya PrakashNo ratings yet
- Alkali SpectraDocument8 pagesAlkali SpectraVivek DewanganNo ratings yet
- CM Homework 3Document5 pagesCM Homework 3mazhariNo ratings yet
- Semiconductor PhotonicsDocument30 pagesSemiconductor PhotonicswuasamomNo ratings yet
- PHY 314: Introduction To Quantum Mechanics, Varsha 2015 Lecture 17 and 18Document8 pagesPHY 314: Introduction To Quantum Mechanics, Varsha 2015 Lecture 17 and 18Ajay KaladharanNo ratings yet
- Degeneracy of Energy Levels of Pseudo-Gaussian Oscillators: Articles You May Be Interested inDocument6 pagesDegeneracy of Energy Levels of Pseudo-Gaussian Oscillators: Articles You May Be Interested inMohammedBujairNo ratings yet
- Lecture 12Document14 pagesLecture 12r prathapNo ratings yet
- Lecture 121111Document26 pagesLecture 121111--No ratings yet
- Molecul h2 NormalisasiDocument5 pagesMolecul h2 NormalisasiendahNo ratings yet
- The Tight Binding Method: 1 Background: A Hierarchy of MethodsDocument15 pagesThe Tight Binding Method: 1 Background: A Hierarchy of MethodsSauvik ChatterjeeNo ratings yet
- Problems 1Document2 pagesProblems 1sodiumcyanide7No ratings yet
- Theory - Classical Harmonic Crystals - Dell.10 12Document24 pagesTheory - Classical Harmonic Crystals - Dell.10 12Bilal Barut100% (1)
- Physics 3 For Electrical Engineering: WWW - Bgu.ac - Il/atomchip WWW - Bgu.ac - Il/nanocenterDocument48 pagesPhysics 3 For Electrical Engineering: WWW - Bgu.ac - Il/atomchip WWW - Bgu.ac - Il/nanocenterJoshNo ratings yet
- Quantum Mechanics HomeworkDocument3 pagesQuantum Mechanics HomeworkJuliana Marquez BustosNo ratings yet
- Qual 19 p2 SolDocument5 pagesQual 19 p2 SolKarishtain NewtonNo ratings yet
- EE335 21 Halfwave Dipole CompressedDocument6 pagesEE335 21 Halfwave Dipole CompressedĐức ĐàoNo ratings yet
- Deuteronlec06 PDFDocument5 pagesDeuteronlec06 PDFAsh LeyNo ratings yet
- Rick Rajter: N N M K 2 I N NDocument4 pagesRick Rajter: N N M K 2 I N Njaurav41No ratings yet
- On The Size of Einsteins Spherically Symmetric UnDocument8 pagesOn The Size of Einsteins Spherically Symmetric UnAqil ShamilNo ratings yet
- (R, θ, z) Φ = Φ (R, z) : spheroidal ellipsoidalDocument28 pages(R, θ, z) Φ = Φ (R, z) : spheroidal ellipsoidalMHSouNo ratings yet
- The Schrödinger Wave Equation For The Hydrogen AtomDocument4 pagesThe Schrödinger Wave Equation For The Hydrogen AtomDannie A. San PedroNo ratings yet
- Current Electricity Sheet SolnsDocument95 pagesCurrent Electricity Sheet SolnsHarsh WardhnNo ratings yet
- Pauli Exclusion PrincipleDocument4 pagesPauli Exclusion PrinciplePedroNo ratings yet
- A Theorem On Unit Regular Rings - T. K. Lee and Y. ZhouDocument6 pagesA Theorem On Unit Regular Rings - T. K. Lee and Y. ZhouSebastian David Higuera RinconNo ratings yet
- Rotation MatrixDocument6 pagesRotation MatrixbenyfirstNo ratings yet
- Ecu 103 Lecture 04Document42 pagesEcu 103 Lecture 04etrial187No ratings yet
- AtomicStrucure SlidesDocument17 pagesAtomicStrucure SlidessamebalutNo ratings yet
- +K2 ArticleDocument14 pages+K2 ArticleFouzia BouchelaghemNo ratings yet
- Hydrogen AtomDocument17 pagesHydrogen AtomTejas SabuNo ratings yet
- 2023 EM1 hw3Document3 pages2023 EM1 hw3810003No ratings yet
- Thanks To Yossef and Shiang Yong For Their Input in This ProblemDocument8 pagesThanks To Yossef and Shiang Yong For Their Input in This ProblemIgnacio JuárezNo ratings yet
- CHAPTER 10: Atomic Structure and Atomic SpectraDocument25 pagesCHAPTER 10: Atomic Structure and Atomic SpectraVijay PradhanNo ratings yet
- The Spectral Theory of Toeplitz Operators. (AM-99), Volume 99From EverandThe Spectral Theory of Toeplitz Operators. (AM-99), Volume 99No ratings yet
- Technical Data: Digital Color Progressive Scan Camera DÜRR TXG06c-K22 - Gigabit EthernetDocument20 pagesTechnical Data: Digital Color Progressive Scan Camera DÜRR TXG06c-K22 - Gigabit EthernetDiogo FiaesNo ratings yet
- UVU Jungle Marathon 2012 BookDocument41 pagesUVU Jungle Marathon 2012 BookGerhard FlatzNo ratings yet
- Problem: Determine The Total Volume of Earth To Be Excavated Up To Elevation 0Document17 pagesProblem: Determine The Total Volume of Earth To Be Excavated Up To Elevation 0gtech00100% (1)
- TPS54160 1.5-A, 60-V, Step-Down DC/DC Converter With Eco-Mode™Document57 pagesTPS54160 1.5-A, 60-V, Step-Down DC/DC Converter With Eco-Mode™sbrhomeNo ratings yet
- EQUILIBRIUMDocument1 pageEQUILIBRIUMMohammed IliasNo ratings yet
- Synchronous Generators: Instructional ObjectivesDocument18 pagesSynchronous Generators: Instructional Objectivessanthosh2009No ratings yet
- Transport Phenomena: τ =μ dv dyDocument2 pagesTransport Phenomena: τ =μ dv dySYED ASGHAR ALI SULTANNo ratings yet
- Louis I KahnDocument27 pagesLouis I KahnKiran BasuNo ratings yet
- Bored Piles - Bilfinger Spezialtiefbau GMBHDocument4 pagesBored Piles - Bilfinger Spezialtiefbau GMBHOga MeoNo ratings yet
- As 1729-1994 Timber - Handles For ToolsDocument7 pagesAs 1729-1994 Timber - Handles For ToolsSAI Global - APACNo ratings yet
- FlapDocument100 pagesFlapRicha Agrawal100% (2)
- Case 621F, 721F Tier 4 Wheel Loader AxelDocument15 pagesCase 621F, 721F Tier 4 Wheel Loader AxelJose CarmonaNo ratings yet
- Lecture 7 - Synchronous Generators 7 PDFDocument28 pagesLecture 7 - Synchronous Generators 7 PDFDorwinNeroNo ratings yet
- Owner: PT. Baker Hughes User: Cok Gede Reza Description: Modified Safety Pin, Add. Handrail & Add. Anti Slip Step Stair at Pressure Test BayDocument3 pagesOwner: PT. Baker Hughes User: Cok Gede Reza Description: Modified Safety Pin, Add. Handrail & Add. Anti Slip Step Stair at Pressure Test BayMuhammad AlpianNo ratings yet
- Absolute Priority Based Cell ReselectionDocument11 pagesAbsolute Priority Based Cell ReselectionNeoRa Ndivo RamsNo ratings yet
- Peniel Integrated Christian Academy of Rizal, Inc. Science Weblinks GRADE 4 S.Y. 2020-2021Document3 pagesPeniel Integrated Christian Academy of Rizal, Inc. Science Weblinks GRADE 4 S.Y. 2020-2021Jhocen Grace GanironNo ratings yet
- HEI Tech Sheet 110Document15 pagesHEI Tech Sheet 110Suganya LokeshNo ratings yet
- Safety Data SheetDocument5 pagesSafety Data SheetAkshay SomaniNo ratings yet
- 02 - D03 - Basic Funtion of SIPROTEC 5Document7 pages02 - D03 - Basic Funtion of SIPROTEC 5DianaNo ratings yet
- New WITTMANN Robots For Large and Small Injection Molding MachinesDocument4 pagesNew WITTMANN Robots For Large and Small Injection Molding MachinesMonark HunyNo ratings yet
- Relative Color Pickup of Three Different Knits and Predictive Dyeing Recipe FormulationDocument17 pagesRelative Color Pickup of Three Different Knits and Predictive Dyeing Recipe FormulationNguyễn Huy CườngNo ratings yet
- SCM and TQM: by Junaid ShaheedDocument8 pagesSCM and TQM: by Junaid ShaheedjunaidsNo ratings yet
- Base On Solid-Works Design of Compact High EfficieDocument15 pagesBase On Solid-Works Design of Compact High EfficieGorgeNo ratings yet
- X PPT CH 12 ElectricityDocument12 pagesX PPT CH 12 ElectricityAakriti100% (1)
- 8210.40 Single Band RET For Multiband Antennas (Controlling White Antenna Array)Document1 page8210.40 Single Band RET For Multiband Antennas (Controlling White Antenna Array)Mohammad AlloushNo ratings yet
- Technical Specification: 1) Filter Feed Pump With Motor 1 NosDocument4 pagesTechnical Specification: 1) Filter Feed Pump With Motor 1 NosKamatchi NathanNo ratings yet
- Ex 4Document4 pagesEx 420-MCE-63 SYED HASSAN KUMAILNo ratings yet
- OHara PDFDocument17 pagesOHara PDFTomás HidalgoNo ratings yet
- BSD-GL-HAL-HMS-100 - (Terms & Definitions)Document42 pagesBSD-GL-HAL-HMS-100 - (Terms & Definitions)Eduard GadzhievNo ratings yet
- ToR For Fiberglass Biogas Plants Installation - EOI PDFDocument4 pagesToR For Fiberglass Biogas Plants Installation - EOI PDFmy09No ratings yet