Download as pdf or txt
You might also like
- Mini Project 4: "Who Was Typing?": EEE350 Dr. Chao Wang Page 1 of 1Document1 pageMini Project 4: "Who Was Typing?": EEE350 Dr. Chao Wang Page 1 of 1ainiNo ratings yet
- Analyzing of Pseudo-Ring Memory Self-Testing Schemes With AlgorithmsDocument8 pagesAnalyzing of Pseudo-Ring Memory Self-Testing Schemes With AlgorithmsijdpsNo ratings yet
- A New Test Pattern Generateor by Altering The Structure of 2-D LFSR For Built in Self Test ApplicationDocument6 pagesA New Test Pattern Generateor by Altering The Structure of 2-D LFSR For Built in Self Test Applicationmobile 1360No ratings yet
- Crawford 2015Document6 pagesCrawford 2015HuyenbobbyNo ratings yet
- Quantifying The Wave-Effect of Irregular LDPC Codes Based On Majority-Based Hard-DecodingDocument5 pagesQuantifying The Wave-Effect of Irregular LDPC Codes Based On Majority-Based Hard-DecodingasdasddsaNo ratings yet
- Expert Systems With Applications: Jing Xiao, Yuping Yan, Jun Zhang, Yong TangDocument8 pagesExpert Systems With Applications: Jing Xiao, Yuping Yan, Jun Zhang, Yong TangmallajosyulasivaniNo ratings yet
- Kanakidis Et Al. - JLT March 2003Document9 pagesKanakidis Et Al. - JLT March 2003myname06No ratings yet
- Turbo-Like Processing For Scalable Interleaving Pattern Generation Application To 60 GHZ UWB-OFDM SystemsDocument6 pagesTurbo-Like Processing For Scalable Interleaving Pattern Generation Application To 60 GHZ UWB-OFDM SystemsPraveen GanigerNo ratings yet
- On Input Selection With Reversible Jump Markov Chain Monte Carlo SamplingDocument10 pagesOn Input Selection With Reversible Jump Markov Chain Monte Carlo SamplingMutahira TahirNo ratings yet
- Cluster Analysis or Clustering Is The Art of Separating The Data Points Into Dissimilar Group With ADocument11 pagesCluster Analysis or Clustering Is The Art of Separating The Data Points Into Dissimilar Group With AramaabbidiNo ratings yet
- This Work Is Supported in Part by The Sensip Center and The Consortium Award No. S0322Document5 pagesThis Work Is Supported in Part by The Sensip Center and The Consortium Award No. S0322Prabhakar EleNo ratings yet
- Artificial Intelligence Methodology For Separation and Classification Partial Discharge SignalsDocument5 pagesArtificial Intelligence Methodology For Separation and Classification Partial Discharge SignalsthetrasancosNo ratings yet
- Dot Plot InterpretationDocument18 pagesDot Plot InterpretationJoão AlvesNo ratings yet
- Analysis of Linear Periodically Time-Varying Circuits by The Frequency Symbolic Method With Applying The D-Trees MethodDocument8 pagesAnalysis of Linear Periodically Time-Varying Circuits by The Frequency Symbolic Method With Applying The D-Trees MethodweylinphilippeNo ratings yet
- A Chaotic Direct-Sequence Spread-Spectrum Communication SystemDocument4 pagesA Chaotic Direct-Sequence Spread-Spectrum Communication SystemNhan BuiNo ratings yet
- Bist Tutorial 2 PDFDocument9 pagesBist Tutorial 2 PDFjcfermosellNo ratings yet
- Gray Box 1994Document10 pagesGray Box 1994Mario CalderonNo ratings yet
- Detect Key Genes in Classification of Microarray DataDocument13 pagesDetect Key Genes in Classification of Microarray DataLeo trinhNo ratings yet
- Gene Sequence AnalysisDocument10 pagesGene Sequence AnalysisÃyûsh KumarNo ratings yet
- Atubriai On Buiikln: SeiktestDocument9 pagesAtubriai On Buiikln: SeiktestVLSISD36 Edwin DominicNo ratings yet
- A Low Complexity VBLAST OFDM Detection Algorithm: Wu Yan, Sumei Sun and Zhongding LeiDocument4 pagesA Low Complexity VBLAST OFDM Detection Algorithm: Wu Yan, Sumei Sun and Zhongding Leibavar88No ratings yet
- Best Linear Unbiased Estimator Approach For Time-Of-Arrival Based LocalisationDocument7 pagesBest Linear Unbiased Estimator Approach For Time-Of-Arrival Based Localisationpks1969No ratings yet
- J Theochem 2004 11 021Document6 pagesJ Theochem 2004 11 021Leonardo VitoriaNo ratings yet
- Signal Processing: Markus Myllyl A, Markku Juntti, Joseph R. CavallaroDocument14 pagesSignal Processing: Markus Myllyl A, Markku Juntti, Joseph R. CavallaroDomRuanNo ratings yet
- Successive Interference Cancellation Via Rank-Reduced Maximum A Posteriori DetectionDocument10 pagesSuccessive Interference Cancellation Via Rank-Reduced Maximum A Posteriori DetectionPawanKumar BarnwalNo ratings yet
- Novel MIMO Detection Algorithm For High-Order Constellations in The Complex DomainDocument14 pagesNovel MIMO Detection Algorithm For High-Order Constellations in The Complex DomainssivabeNo ratings yet
- Neighboring Cell Search Techniques For LTE Systems: CitationDocument7 pagesNeighboring Cell Search Techniques For LTE Systems: Citationacela3248No ratings yet
- Iterative Receivers Combining MIMO DetectionDocument19 pagesIterative Receivers Combining MIMO Detectionrida4No ratings yet
- Supporting Information: Supplementary Note 1 Encoding: Data To DNA SequencesDocument25 pagesSupporting Information: Supplementary Note 1 Encoding: Data To DNA Sequencesweixing wangNo ratings yet
- An Alternative Approach For Generation of Membership Functions and Fuzzy Rules Based On Radial and Cubic Basis Function NetworksDocument20 pagesAn Alternative Approach For Generation of Membership Functions and Fuzzy Rules Based On Radial and Cubic Basis Function NetworksAntonio Madueño LunaNo ratings yet
- EM Ver7Document5 pagesEM Ver7Theodoros TsiftsisNo ratings yet
- Comparison of Two Novel List Sphere Detector Algorithms For Mimo-Ofdm SystemsDocument5 pagesComparison of Two Novel List Sphere Detector Algorithms For Mimo-Ofdm SystemsDomRuanNo ratings yet
- 12 2 4 PDFDocument4 pages12 2 4 PDFDyanaHottest-GirlDavidsonNo ratings yet
- Formato Proyecto I+DDocument3 pagesFormato Proyecto I+DVanessa Fontalvo renizNo ratings yet
- Ashwani Singh 117BM0731Document28 pagesAshwani Singh 117BM0731Ashwani SinghNo ratings yet
- Efficiency Ordering of StochasDocument14 pagesEfficiency Ordering of StochasTom SmithNo ratings yet
- Combined Fuzzy-Logic Wavelet-Based Fault Classification Technique For Power System RelayingDocument8 pagesCombined Fuzzy-Logic Wavelet-Based Fault Classification Technique For Power System Relayingوليد موسىNo ratings yet
- 09-03-03 PDCCH Power BoostingDocument5 pages09-03-03 PDCCH Power BoostingANDRETERROMNo ratings yet
- Iterative Autoassociative Memory Models For Image Recalls and PaDocument6 pagesIterative Autoassociative Memory Models For Image Recalls and Pasathiya vrsNo ratings yet
- Phase Shifter MergingDocument2 pagesPhase Shifter Mergingeswar chandNo ratings yet
- Bde DbscanDocument11 pagesBde DbscanBruno Romero de AzevedoNo ratings yet
- Title: Status: Purpose: Author(s) or Contact(s)Document4 pagesTitle: Status: Purpose: Author(s) or Contact(s)Shafayet UddinNo ratings yet
- Algorithmica: Sorting-Based Selection Algorithms For Hypercubic NetworksDocument18 pagesAlgorithmica: Sorting-Based Selection Algorithms For Hypercubic NetworksasddsdsaNo ratings yet
- Tmp939a TMPDocument17 pagesTmp939a TMPFrontiersNo ratings yet
- Finite-State Markov Modeling of Correlated Rician-Fading ChannelsDocument11 pagesFinite-State Markov Modeling of Correlated Rician-Fading ChannelsIamguesto NickNo ratings yet
- Physical Communication: Ameen Abdelmutalab Khaled Assaleh Mohamed El-TarhuniDocument9 pagesPhysical Communication: Ameen Abdelmutalab Khaled Assaleh Mohamed El-TarhuniAsil AsilaNo ratings yet
- Classifier Based Text Mining For Radial Basis FunctionDocument6 pagesClassifier Based Text Mining For Radial Basis FunctionEdinilson VidaNo ratings yet
- Plagiarism Checker X Originality: Similarity Found: 22%Document9 pagesPlagiarism Checker X Originality: Similarity Found: 22%alisha DhawanNo ratings yet
- Kernel Nearest-Neighbor AlgorithmDocument10 pagesKernel Nearest-Neighbor AlgorithmDebora OlivaresNo ratings yet
- Cho Icassp2019 08683380-1Document5 pagesCho Icassp2019 08683380-1reschyNo ratings yet
- Physical Layer Deep Learning of Encodings For The MIMO Fading ChannelDocument5 pagesPhysical Layer Deep Learning of Encodings For The MIMO Fading Channeljun zhaoNo ratings yet
- Encryption and Decryption Algorithm Using Two Dimensional Cellular Automata Rules in CryptographyDocument6 pagesEncryption and Decryption Algorithm Using Two Dimensional Cellular Automata Rules in CryptographybhasinshresthNo ratings yet
- Delay Extraction-Based Passive Macromodeling Techniques For Transmission Line Type Interconnects Characterized by Tabulated Multiport DataDocument13 pagesDelay Extraction-Based Passive Macromodeling Techniques For Transmission Line Type Interconnects Characterized by Tabulated Multiport DataPramod SrinivasanNo ratings yet
- Neural Network Model Predictive Control of Nonlinear Systems Using Genetic AlgorithmsDocument10 pagesNeural Network Model Predictive Control of Nonlinear Systems Using Genetic Algorithmszaihry fuentesNo ratings yet
- A Study On Reconstruction of Linear Scrambler Using Dual Words of Channel EncoderDocument11 pagesA Study On Reconstruction of Linear Scrambler Using Dual Words of Channel EncoderlapbinNo ratings yet
- A Relational Approach To The Compilation of Sparse Matrix ProgramsDocument16 pagesA Relational Approach To The Compilation of Sparse Matrix Programsmmmmm1900No ratings yet
- Comparative Study - BSPDocument4 pagesComparative Study - BSPNayana RaoNo ratings yet
- Poster Template Compbiotech Diaz-Carral v3 1-4-1Document1 pagePoster Template Compbiotech Diaz-Carral v3 1-4-1Ángel Díaz CarralNo ratings yet
- Throughput and Error State Performance of AMC Scheme in 3G Wireless SystemsDocument4 pagesThroughput and Error State Performance of AMC Scheme in 3G Wireless SystemsInternational Organization of Scientific Research (IOSR)No ratings yet
- Next Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsFrom EverandNext Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsNo ratings yet
- Spline and Spline Wavelet Methods with Applications to Signal and Image Processing: Volume III: Selected TopicsFrom EverandSpline and Spline Wavelet Methods with Applications to Signal and Image Processing: Volume III: Selected TopicsNo ratings yet
- Circle Geometry PowerpointDocument68 pagesCircle Geometry PowerpointKadhirvelou Dhandapani100% (2)
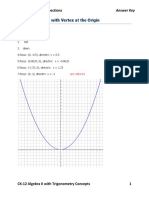
- 10.1 Parabolas With Vertex at The Origin: Chapter 10 - Conic SectionsDocument28 pages10.1 Parabolas With Vertex at The Origin: Chapter 10 - Conic SectionsShanelle SantillanaNo ratings yet
- What Is Angle Bisector TheoremDocument8 pagesWhat Is Angle Bisector TheoremTabish RozaanNo ratings yet
- 12th Maths EngMed QueBank MSCERTDocument103 pages12th Maths EngMed QueBank MSCERTmr. MorningstarNo ratings yet
- Cohen A.M. - Numerical Methods For Laplace Transform Inversion - Program Code Only (2007)Document57 pagesCohen A.M. - Numerical Methods For Laplace Transform Inversion - Program Code Only (2007)All EducateNo ratings yet
- CH 08Document48 pagesCH 08KavunNo ratings yet
- Boyce ODEch 2 S MP 21Document2 pagesBoyce ODEch 2 S MP 21Elza Dwi PutriNo ratings yet
- C2 - Simplifying Logic Circuits Using Theorems-NTHai-2-2020Document6 pagesC2 - Simplifying Logic Circuits Using Theorems-NTHai-2-2020Duc NguyenNo ratings yet
- Modern Algebra (CH 10, Rings 1) RA Maths Test 27.5.2018Document2 pagesModern Algebra (CH 10, Rings 1) RA Maths Test 27.5.2018M Murugan100% (1)
- Lec 8 - Polish Notation 24102022 101228amDocument17 pagesLec 8 - Polish Notation 24102022 101228amdoodhjaleybiNo ratings yet
- Vietnam IMO Booklet 2017Document37 pagesVietnam IMO Booklet 2017garciacapitanNo ratings yet
- A Proof of The Kahn-Kalai Conjecture: Jinyoung Park and Huy Tuan PhamDocument6 pagesA Proof of The Kahn-Kalai Conjecture: Jinyoung Park and Huy Tuan PhamScribd MusabeNo ratings yet
- Straight Lines 6-Parallel PerpendicularDocument10 pagesStraight Lines 6-Parallel PerpendicularSelva RajNo ratings yet
- Personal Statement (Sample)Document2 pagesPersonal Statement (Sample)Justin Folk100% (1)
- Moore 1968Document3 pagesMoore 1968Leqia HeNo ratings yet
- Department of Software Engineering Programming Fundamentals-1Document2 pagesDepartment of Software Engineering Programming Fundamentals-1AsadNo ratings yet
- Relational Algebra and Relational Calculus: in The Course Textbook Domain and Tuple Relational Calculus Ramesh@cs - Ubc.caDocument47 pagesRelational Algebra and Relational Calculus: in The Course Textbook Domain and Tuple Relational Calculus Ramesh@cs - Ubc.casrikanth anumaNo ratings yet
- Math 23 Lecture 3.2 More On Triple Integrals PDFDocument19 pagesMath 23 Lecture 3.2 More On Triple Integrals PDFyeahNo ratings yet
- Topic 5 CompleteDocument34 pagesTopic 5 CompleteWasif AhmedNo ratings yet
- Fundamental Finite Element Analysis and ApplicationsDocument718 pagesFundamental Finite Element Analysis and Applicationschamanchandel79% (28)
- Counting With Permutations and Combinations CK-1Document12 pagesCounting With Permutations and Combinations CK-1Ratan KumawatNo ratings yet
- Operations ResearchDocument208 pagesOperations ResearchGokul Chandrappa0% (1)
- CTQ Nimcet 2022: Straight Line IDocument3 pagesCTQ Nimcet 2022: Straight Line INetwork BusyNo ratings yet
- 133 Week2Document18 pages133 Week2E_dienNo ratings yet
- MMW Semi Final ExamDocument2 pagesMMW Semi Final ExamSabiniano Kris MellanNo ratings yet
- Color Image Enhancement Using The Support Fuzzification in The FrameworkDocument40 pagesColor Image Enhancement Using The Support Fuzzification in The FrameworkCory HornNo ratings yet
- Mathswatch Higher Worksheets Aw PDFDocument147 pagesMathswatch Higher Worksheets Aw PDF가예은No ratings yet
- 2021 Output2 MathadvanceDocument6 pages2021 Output2 MathadvanceJeffrey JonasNo ratings yet
- Formula - XM f2Document14 pagesFormula - XM f2Nurul Binti YusofNo ratings yet