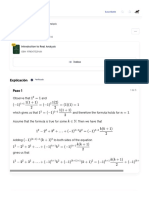
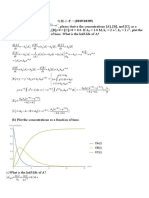
Download as pdf or txt
You might also like
- Test Bank For Discrete Mathematics With Applications 5th Edition Susanna S EppDocument22 pagesTest Bank For Discrete Mathematics With Applications 5th Edition Susanna S Eppernestocolonx88p100% (17)
- Lectures On Quantum Mechanics: Giuseppe E. SantoroDocument90 pagesLectures On Quantum Mechanics: Giuseppe E. SantoroTudor PatuleanuNo ratings yet
- Lecture 13Document19 pagesLecture 13hema maliniNo ratings yet
- Ejercicio 5Document3 pagesEjercicio 5Johanni Paniagua De La CruzNo ratings yet
- Lecture3 (Adsorption-Part2) 2022Document8 pagesLecture3 (Adsorption-Part2) 2022최종윤No ratings yet
- Wahyu Dan Inu (3.32 Dan 3.36)Document3 pagesWahyu Dan Inu (3.32 Dan 3.36)Indah pratiwiNo ratings yet
- Topic Wise Test-6 Band Theory Of-Solid (Solution)Document7 pagesTopic Wise Test-6 Band Theory Of-Solid (Solution)Řůpäm ŔøýNo ratings yet
- Semiconductor Electronic StructureDocument46 pagesSemiconductor Electronic StructureXiao AlexNo ratings yet
- Ps8sol 6245 2004Document6 pagesPs8sol 6245 2004Tejas PatilNo ratings yet
- NPTEL Online Course: Control Engineering: Assignment 1Document4 pagesNPTEL Online Course: Control Engineering: Assignment 1udayNo ratings yet
- x1 (K) +x2 (K) +u (K)Document1 pagex1 (K) +x2 (K) +u (K)Nãčėr SebaâNo ratings yet
- Ex1 Qu2Document2 pagesEx1 Qu2Manasa YNo ratings yet
- Lecture 1Document8 pagesLecture 1sabbithiNo ratings yet
- 108 Quiz1Document1 page108 Quiz1沈智恩No ratings yet
- Chapter 14. Enzyme KineticsDocument44 pagesChapter 14. Enzyme KineticsKalai VaniNo ratings yet
- MATH1025 Number Systems Homework Problems 2Document4 pagesMATH1025 Number Systems Homework Problems 2bananacompNo ratings yet
- Electronic Journal of Combinatorial Number TheoryDocument17 pagesElectronic Journal of Combinatorial Number TheoryRalu DumiNo ratings yet
- Perturbation BasicsDocument3 pagesPerturbation Basicsresat akanNo ratings yet
- Homework 14 Colley 7 3Document5 pagesHomework 14 Colley 7 3William BradleyNo ratings yet
- EE402 Lecture 3Document5 pagesEE402 Lecture 3sdfgNo ratings yet
- Physics 385 Assignment 7: Problem 8-3Document5 pagesPhysics 385 Assignment 7: Problem 8-3wizbizphdNo ratings yet
- K - K Versus K - (k+1) - (E)Document4 pagesK - K Versus K - (k+1) - (E)Gabriel BerceNo ratings yet
- 2011-01-Elliptic Functions - Complex Variables!!!!!!within-Jacobi's Imaginary TransformationDocument7 pages2011-01-Elliptic Functions - Complex Variables!!!!!!within-Jacobi's Imaginary Transformationdawson zhaoNo ratings yet
- The Hamiltonian Analysis of Lattice Vibrations. Phononic BandgapDocument6 pagesThe Hamiltonian Analysis of Lattice Vibrations. Phononic BandgapgabrielNo ratings yet
- Kronnig-Penney Using Block TheoremDocument6 pagesKronnig-Penney Using Block TheoremChris EvanNo ratings yet
- MMSB Exercise SolutionsDocument49 pagesMMSB Exercise SolutionsYolanda Winarny Eviphanie HutabaratNo ratings yet
- Structural Dynamics: 2CE611 by DR S S Mishra, NIT Patna: Rayleigh QuotientDocument7 pagesStructural Dynamics: 2CE611 by DR S S Mishra, NIT Patna: Rayleigh QuotientRoshni TNo ratings yet
- 1998 99 Prob Solving ps2Document13 pages1998 99 Prob Solving ps2biswacementNo ratings yet
- Derivation of Fresnel DiffractionDocument5 pagesDerivation of Fresnel DiffractionWhalsy nNo ratings yet
- Schaum's College Algebra - Ch. 31Document60 pagesSchaum's College Algebra - Ch. 31JNo ratings yet
- Lecture43 - Page286 - Ratio TestDocument3 pagesLecture43 - Page286 - Ratio TestViral TouchNo ratings yet
- Vibrations: Free Response of Multi-Degree-of-Freedom SystemsDocument8 pagesVibrations: Free Response of Multi-Degree-of-Freedom SystemsCharlie TejNo ratings yet
- Chapter 2. Homogeneous SemiconductorsDocument49 pagesChapter 2. Homogeneous Semiconductorsfourier76No ratings yet
- Logan 6e ISM Chapter 02Document22 pagesLogan 6e ISM Chapter 02leehyobeenNo ratings yet
- A) - Equilibrium Approach: K K K K K K ES) SDocument6 pagesA) - Equilibrium Approach: K K K K K K ES) Salfi hendryNo ratings yet
- 8.simple Harmonic Motion and ElasticityExercise PDFDocument81 pages8.simple Harmonic Motion and ElasticityExercise PDFSubbalakshmi Yeleswarapu 010311100% (1)
- Compressible Flow Tut AddDocument2 pagesCompressible Flow Tut AddPeter AdamNo ratings yet
- CIVE50003 Computational Methods II - Lecture VI - 030223 V2Document27 pagesCIVE50003 Computational Methods II - Lecture VI - 030223 V2TwinyNo ratings yet
- Discrete Mathematics and Its Applications - 9780073383095 - Exercise 19 - QuizletDocument1 pageDiscrete Mathematics and Its Applications - 9780073383095 - Exercise 19 - QuizletTime DilationNo ratings yet
- PRACTICE EXERCISES IMCSIngaporeDocument6 pagesPRACTICE EXERCISES IMCSIngaporeJoshRobertD.ObaobNo ratings yet
- CDROM-Appendix-K FoglerDocument3 pagesCDROM-Appendix-K FoglerHenryVladimirDelgadoCanaviriNo ratings yet
- Solution Manual - Chapter 22-23: Jens Zamanian May 7, 2014Document8 pagesSolution Manual - Chapter 22-23: Jens Zamanian May 7, 2014Gilvan PirocaNo ratings yet
- Mathematical InductionDocument17 pagesMathematical Inductionmita sari halawaNo ratings yet
- Test Practice Exam Questions and Answers Solow ModelDocument5 pagesTest Practice Exam Questions and Answers Solow ModelFiranbek BgNo ratings yet
- Exercise MPC1Document16 pagesExercise MPC1Ifrah AlamNo ratings yet
- AssignmentDocument5 pagesAssignmentharshitsharma.siwalNo ratings yet
- Full Download First Course in The Finite Element Method 6th Edition Logan Solutions ManualDocument36 pagesFull Download First Course in The Finite Element Method 6th Edition Logan Solutions Manualmariputwashercy4ca100% (40)
- Dwnload Full First Course in The Finite Element Method 6th Edition Logan Solutions Manual PDFDocument36 pagesDwnload Full First Course in The Finite Element Method 6th Edition Logan Solutions Manual PDFcostyrafic2100% (13)
- 09 Golnaraghi App I I-1-I-2Document2 pages09 Golnaraghi App I I-1-I-2Adam KhanNo ratings yet
- Fourier 401Document10 pagesFourier 401Doglasse João MárioNo ratings yet
- Exercise 2: Convolution: EG1110 Signals and SystemsDocument5 pagesExercise 2: Convolution: EG1110 Signals and Systemssrinvas_107796724No ratings yet
- Basic Matrix Operations: - TransposeDocument26 pagesBasic Matrix Operations: - Transposesaraei7142356No ratings yet
- ( T, (M) (O) F: Motion in Fields Exercise 1Document6 pages( T, (M) (O) F: Motion in Fields Exercise 1Lionel TebonNo ratings yet
- Electrostatistics (Practice Questions PDFDocument32 pagesElectrostatistics (Practice Questions PDFSakshamNo ratings yet
- Full Download First Course in The Finite Element Method Si Edition 6th Edition Logan Solutions ManualDocument36 pagesFull Download First Course in The Finite Element Method Si Edition 6th Edition Logan Solutions Manualmariputwashercy4ca98% (42)
- Dwnload Full First Course in The Finite Element Method Si Edition 6th Edition Logan Solutions Manual PDFDocument36 pagesDwnload Full First Course in The Finite Element Method Si Edition 6th Edition Logan Solutions Manual PDFcostyrafic2100% (17)
- Exercico 5.6 ListaDocument4 pagesExercico 5.6 ListaSara AvizNo ratings yet
- PropagateDocument11 pagesPropagateMichel Rodrigues AndradeNo ratings yet
- 5.62 Physical Chemistry Ii: Mit OpencoursewareDocument6 pages5.62 Physical Chemistry Ii: Mit OpencoursewareavdvNo ratings yet
- 5.62 Physical Chemistry Ii: Mit OpencoursewareDocument10 pages5.62 Physical Chemistry Ii: Mit OpencoursewareavdvNo ratings yet
- 5.62 Physical Chemistry Ii: Mit OpencoursewareDocument6 pages5.62 Physical Chemistry Ii: Mit OpencoursewareavdvNo ratings yet
- 5.62 Physical Chemistry Ii: Mit OpencoursewareDocument8 pages5.62 Physical Chemistry Ii: Mit OpencoursewareavdvNo ratings yet
- 5.62 Physical Chemistry Ii: Mit OpencoursewareDocument10 pages5.62 Physical Chemistry Ii: Mit OpencoursewareavdvNo ratings yet
- 5.62 Physical Chemistry Ii: Mit OpencoursewareDocument17 pages5.62 Physical Chemistry Ii: Mit OpencoursewareavdvNo ratings yet
- 5.62 Physical Chemistry Ii: Mit OpencoursewareDocument8 pages5.62 Physical Chemistry Ii: Mit OpencoursewareavdvNo ratings yet
- Scanned by CamscannerDocument3 pagesScanned by CamscanneravdvNo ratings yet
- Iapt Junior Science-2017: Student Registration FormDocument1 pageIapt Junior Science-2017: Student Registration FormavdvNo ratings yet
- Molecular Geometry and Bonding TheoriesDocument5 pagesMolecular Geometry and Bonding TheoriesPineraserNo ratings yet
- Hallwachs - Poster-A4-EnDocument1 pageHallwachs - Poster-A4-Enapi-281473337No ratings yet
- Eeer16 PDFDocument143 pagesEeer16 PDFRayapati Devi PrasadNo ratings yet
- The Potential and Challenges of Time-Resolved SingDocument33 pagesThe Potential and Challenges of Time-Resolved SingJosh GonzalesNo ratings yet
- Clebsch Gordan CoefficientsDocument1 pageClebsch Gordan CoefficientsLangleiFanNo ratings yet
- Chp07ans PDFDocument28 pagesChp07ans PDFBinit KarNo ratings yet
- Photoelectron Spectroscopy, ARPES and PED - PrinciplesDocument62 pagesPhotoelectron Spectroscopy, ARPES and PED - PrinciplesVlad NeacsuNo ratings yet
- Toward A Scientific, Sacred Indology: Prof. Ravi Gomatam (Aka Rasaraja Dasa)Document21 pagesToward A Scientific, Sacred Indology: Prof. Ravi Gomatam (Aka Rasaraja Dasa)gaurnityanandaNo ratings yet
- 202006151236284892NK-Properties of Laser BeamDocument5 pages202006151236284892NK-Properties of Laser BeamJatin botNo ratings yet
- Quantum Physics Lecture NotesDocument43 pagesQuantum Physics Lecture NotesSwayangdipta10 16208No ratings yet
- Indian Institute of Technology DelhiDocument5 pagesIndian Institute of Technology DelhiAchyut SalunkheNo ratings yet
- Class 11 Chemistry Important Question Answer of Chapter 2 (Structure of Atom) &chapter 3 (Classification of Elements and Periodicity in PropertiesDocument38 pagesClass 11 Chemistry Important Question Answer of Chapter 2 (Structure of Atom) &chapter 3 (Classification of Elements and Periodicity in Propertiessriram.j.athreyaNo ratings yet
- Quantum Mechanics PDFDocument49 pagesQuantum Mechanics PDFIjack HrangchalNo ratings yet
- Why Mercury Liquid?: Or, Why Do Relativistic Effects Not Get Into Chemistry Textbooks?Document4 pagesWhy Mercury Liquid?: Or, Why Do Relativistic Effects Not Get Into Chemistry Textbooks?leonardo_strajaneliNo ratings yet
- Iffraction Refers To Various Phenomena That Occur When ADocument9 pagesIffraction Refers To Various Phenomena That Occur When ArodolfoccrNo ratings yet
- Notes 20Document5 pagesNotes 20dhy7rnphy6No ratings yet
- Science 9 Quarter 2 Module 1Document31 pagesScience 9 Quarter 2 Module 1Amara KimNo ratings yet
- Gravitation - Frequency Shift Vs LensingDocument3 pagesGravitation - Frequency Shift Vs LensingPierre NICOLENo ratings yet
- Chem 1301 - 2021 MidtermDocument14 pagesChem 1301 - 2021 MidtermRandom PersonNo ratings yet
- Grade 9 Science Chemistry 1 DLPDocument13 pagesGrade 9 Science Chemistry 1 DLPManongdo AllanNo ratings yet
- Introduction To The Quantum Theory (2nd Ed) - David ParkDocument3 pagesIntroduction To The Quantum Theory (2nd Ed) - David Parkmach20_aardvark8064No ratings yet
- Assignment-3 - LS Coupling and JJ CouplingDocument3 pagesAssignment-3 - LS Coupling and JJ Couplingm9461309595No ratings yet
- Advanced Atomic StructureDocument6 pagesAdvanced Atomic Structurek_chilukuriNo ratings yet
- Statistical MechanicsDocument15 pagesStatistical MechanicsSadhin SaleemNo ratings yet
- Tugas Fisika IntiDocument35 pagesTugas Fisika IntiSharasanty PNo ratings yet
- Physics of Spin (10 Points) Part A. Larmor Precession (1.6 Points)Document3 pagesPhysics of Spin (10 Points) Part A. Larmor Precession (1.6 Points)Khongor DamdinbayarNo ratings yet
- Electron Configuration Study SheetDocument3 pagesElectron Configuration Study SheetmyllenaNo ratings yet
- Particle Interactions 1 - A LevelDocument16 pagesParticle Interactions 1 - A LevelAdebayo WalidNo ratings yet
- Quantum Mechanics IsDocument2 pagesQuantum Mechanics IsPadayao, Martin Jan C.No ratings yet