
Like Disolve Like
Like Disolve Like
You might also like
- Advanced Acids and BasesDocument13 pagesAdvanced Acids and BasesJohn Carlo MacalagayNo ratings yet
- Exercise 1 and 2 - Revised2Document5 pagesExercise 1 and 2 - Revised2Vivekka Olivia JohnNo ratings yet
- 4 Heat Exchanger 2Document42 pages4 Heat Exchanger 2januari desemberNo ratings yet
- WjceDocument5 pagesWjceshmohiuddinNo ratings yet
- Flatt Ve ArkDocument6 pagesFlatt Ve ArkMert GüngörNo ratings yet
- GaslawconstantDocument6 pagesGaslawconstantSefa Ceren KANDEMİRNo ratings yet
- Determination of The Molecular Weight of A Non-Volatile Solid by The Cryoscopic MethodDocument13 pagesDetermination of The Molecular Weight of A Non-Volatile Solid by The Cryoscopic Methodariana religiosoNo ratings yet
- Steam Z FactorDocument7 pagesSteam Z Factorralf.stein9265No ratings yet
- Ie13 2 0170Document6 pagesIe13 2 0170jnwxNo ratings yet
- Ubc 1999-464040Document235 pagesUbc 1999-464040Pavita SalsabilaNo ratings yet
- Assessment of Calculation Methods For Calcium Carbonate Saturation in Drinking Water For DIN 38404-10 ComplianceDocument10 pagesAssessment of Calculation Methods For Calcium Carbonate Saturation in Drinking Water For DIN 38404-10 ComplianceinejattNo ratings yet
- Calculation of Solvation Free Energies of Charged Solutes Using Mixed Cluster/Continuum ModelsDocument11 pagesCalculation of Solvation Free Energies of Charged Solutes Using Mixed Cluster/Continuum ModelsMohon GuptaNo ratings yet
- Ba Doped Co POMDocument7 pagesBa Doped Co POMpandiaraj1988No ratings yet
- Calculation of Phase Diagrams of Gas-HydratesDocument9 pagesCalculation of Phase Diagrams of Gas-HydratesMichael ParkerNo ratings yet
- Uyfrbfhrb 2Document19 pagesUyfrbfhrb 2Bill644No ratings yet
- Hay Duk 1982Document5 pagesHay Duk 1982FSBollNo ratings yet
- RyznarDocument12 pagesRyznarJim FrenkenNo ratings yet
- X 2 G 63 TSANDOVALDocument13 pagesX 2 G 63 TSANDOVALChemical Engineering Dance CrewNo ratings yet
- Sifat KoligatifDocument12 pagesSifat Koligatiflilik supriantiNo ratings yet
- Characterization of Partially Hydrolyzed Poly (Vinyl Alcohol) - Effect of Poly (Vinyl Alcohol) Molecular Architecture On Aqueous Phase ConformationDocument8 pagesCharacterization of Partially Hydrolyzed Poly (Vinyl Alcohol) - Effect of Poly (Vinyl Alcohol) Molecular Architecture On Aqueous Phase ConformationIrvan YudhistiraNo ratings yet
- Water Index CalculationsDocument24 pagesWater Index CalculationsGustavo Adolfo Piñero Borges100% (1)
- Humberto Hugo Soriano ValdiviaDocument12 pagesHumberto Hugo Soriano ValdiviaBeto SorianoNo ratings yet
- CangrumaDocument10 pagesCangrumaAna ChisNo ratings yet
- 6211 METHANEDocument3 pages6211 METHANERuth Coronado ChuyesNo ratings yet
- Ilcpa 5 20Document15 pagesIlcpa 5 20widya nurul ainiNo ratings yet
- Ion Exchange Report - Group 4 Section 2Document18 pagesIon Exchange Report - Group 4 Section 2razan.tarabay.26No ratings yet
- 1 s2.0 S1876610211005960 MainDocument8 pages1 s2.0 S1876610211005960 MainRenalyn TorioNo ratings yet
- Prediction of Scale Problems Due To Injection of Incompatible WatersDocument12 pagesPrediction of Scale Problems Due To Injection of Incompatible WatersMiguel GrNo ratings yet
- Uyfrbfhrb 3Document18 pagesUyfrbfhrb 3Bill644No ratings yet
- Cheng 2013Document10 pagesCheng 2013Cristian CastilloNo ratings yet
- A Compositional Model For CO2 Floods Including CO2 Solubility in WaterDocument13 pagesA Compositional Model For CO2 Floods Including CO2 Solubility in Watermoji20067147No ratings yet
- Chapter7 PDFDocument36 pagesChapter7 PDFnillnjtNo ratings yet
- Practica ConductimetriaDocument21 pagesPractica Conductimetriajoss villagomezNo ratings yet
- Bord PaperkorrDocument9 pagesBord Paperkorrjakalae5263No ratings yet
- EXP2 Molar Mass of A Volatile Liquid Final ReportDocument14 pagesEXP2 Molar Mass of A Volatile Liquid Final ReportBINSAHNo ratings yet
- 10.1021 Je034179fDocument5 pages10.1021 Je034179faminbm.pt24No ratings yet
- Paper No.: Corrosion Behavior of Carbon Steel in Supercritical Co - Water EnvironmentsDocument20 pagesPaper No.: Corrosion Behavior of Carbon Steel in Supercritical Co - Water Environmentss arvelakisNo ratings yet
- رێلوەه-نیددەحلاەس یۆکناز Salahaddin University-Erbil: (solubility product constant)Document8 pagesرێلوەه-نیددەحلاەس یۆکناز Salahaddin University-Erbil: (solubility product constant)shko noshaNo ratings yet
- Jurnal 5 - (Cox Et Al., 2015 - Henry Constant ReferenceDocument13 pagesJurnal 5 - (Cox Et Al., 2015 - Henry Constant Referenceanik suciNo ratings yet
- Catalysts: New Method of Determining Kinetic Parameters For Decomposition of Hydrogen Peroxide by CatalaseDocument12 pagesCatalysts: New Method of Determining Kinetic Parameters For Decomposition of Hydrogen Peroxide by CatalaseSo PhiaNo ratings yet
- IP 3. Protocol - Chemical Principles II LaboratoryDocument9 pagesIP 3. Protocol - Chemical Principles II LaboratoryJavier PratdesabaNo ratings yet
- Chemistry LaboratoryDocument31 pagesChemistry LaboratoryBunty KhiljiNo ratings yet
- Softening: Drinking Water - Lab ExperimentsDocument6 pagesSoftening: Drinking Water - Lab Experimentsnermeen ahmedNo ratings yet
- Kinetics of Iodide-Catalyzed Decomposition of Hydrogen Peroxide in A Continuous-Stirred Tank ReactorDocument6 pagesKinetics of Iodide-Catalyzed Decomposition of Hydrogen Peroxide in A Continuous-Stirred Tank ReactorGlen TrinidadNo ratings yet
- Reservoir Simulation ModelDocument6 pagesReservoir Simulation ModelDeejayF1No ratings yet
- Solubility Measurement and Modeling For PropaneDocument8 pagesSolubility Measurement and Modeling For PropanerschirtNo ratings yet
- Datos IsopiésticoDocument4 pagesDatos IsopiésticoJuanMeMooMillaNo ratings yet
- Improving The Hydrogen Oxidation Reaction Rate by Promotion of Hydroxyl AdsorptionDocument7 pagesImproving The Hydrogen Oxidation Reaction Rate by Promotion of Hydroxyl AdsorptionHasanulKamilNo ratings yet
- 0437 0454 PDFDocument18 pages0437 0454 PDFzhyhhNo ratings yet
- 0437 0454 PDFDocument18 pages0437 0454 PDFzhyhhNo ratings yet
- Thermodynamic Models For Determination of The Solubility of F2CDocument6 pagesThermodynamic Models For Determination of The Solubility of F2Ctatchanok1525No ratings yet
- Glycol Dehydration of High-Acid Gas StreamsDocument8 pagesGlycol Dehydration of High-Acid Gas StreamsAqsam NaveedNo ratings yet
- Sample Chemistry Undergraduate Laboratory ReportDocument14 pagesSample Chemistry Undergraduate Laboratory ReportApril TapayanNo ratings yet
- 1 s2.0 S001346862101999X MainDocument13 pages1 s2.0 S001346862101999X MainROBSON CardosoNo ratings yet
- Energies 17 02356 v2Document39 pagesEnergies 17 02356 v2Niraj ThakreNo ratings yet
- PH Meter It's Applications: Practical Advance Pharmaceutical AnalysisDocument19 pagesPH Meter It's Applications: Practical Advance Pharmaceutical AnalysisarjunkumbharchemNo ratings yet
- 00071326Document12 pages00071326theoneandonly1No ratings yet
- Caoh2 3rd EdDocument9 pagesCaoh2 3rd Edcano96No ratings yet
- SPE-203762-MS Using Difference Factor Thermodynamics Models in Hydrate Formation Prediction During Surface Production Well TestingDocument14 pagesSPE-203762-MS Using Difference Factor Thermodynamics Models in Hydrate Formation Prediction During Surface Production Well TestingTheNourEldenNo ratings yet
- Water Rock InteractionDocument4 pagesWater Rock InteractionPritam RajNo ratings yet
- Zhang 2014Document7 pagesZhang 2014Jordy Romario Grados SoriaNo ratings yet
- Steam TracingDocument18 pagesSteam TracingSyed Mujtaba Ali Bukhari100% (2)
- Us 5221800Document7 pagesUs 5221800Mochamad Abdul MalikNo ratings yet
- Colorkote: Technical Data SheetDocument2 pagesColorkote: Technical Data Sheet김종태No ratings yet
- TEX - CHEM 103 Organic ChemistryDocument52 pagesTEX - CHEM 103 Organic ChemistrychioNo ratings yet
- Perfringens Agar Base (Tryptose Sulphite Cycloserine Agar Base) (Iso 7937 2004, Iso 14189 2013) - TD-TM 1826Document3 pagesPerfringens Agar Base (Tryptose Sulphite Cycloserine Agar Base) (Iso 7937 2004, Iso 14189 2013) - TD-TM 1826Ventas LabsupplyNo ratings yet
- Prob11 25Document1 pageProb11 25ozi125_2No ratings yet
- EN-GJS-450-10, ASTM A536 65-45-12, Ductile Iron, SG Iron Chemical Composition, Mechanical and Physical PropertiesDocument2 pagesEN-GJS-450-10, ASTM A536 65-45-12, Ductile Iron, SG Iron Chemical Composition, Mechanical and Physical PropertiesBbiettg AndiarNo ratings yet
- Ficha Tecnica Compresor FiacDocument10 pagesFicha Tecnica Compresor FiacSAUL OSPINONo ratings yet
- Petroleum Exploration MetDocument10 pagesPetroleum Exploration MetShiraz NajatNo ratings yet
- E7010 P1 - 15680891Document4 pagesE7010 P1 - 15680891Arvind SahaniNo ratings yet
- Biosurfactant 7Document8 pagesBiosurfactant 7Aranrie MosesNo ratings yet
- Selection Control ValvesDocument7 pagesSelection Control ValvesVeera Mani100% (1)
- EXPLOSIVE CLASSIFICATIONS ATexDocument5 pagesEXPLOSIVE CLASSIFICATIONS ATexJeet SinghNo ratings yet
- Ez-10 MSDSDocument5 pagesEz-10 MSDSMark Evan SalutinNo ratings yet
- Syltherm 800 Silicone Heat Transfer Fluid MSDSDocument8 pagesSyltherm 800 Silicone Heat Transfer Fluid MSDSNasser AlshalwyNo ratings yet
- Acs Jchemed 7b00739 PDFDocument9 pagesAcs Jchemed 7b00739 PDFDaniel Ernesto Ruiz MondragonNo ratings yet
- Reflex Plus: The Powder Solve ApproachDocument4 pagesReflex Plus: The Powder Solve Approachmetal2567No ratings yet
- Si-COAT® 579-SDSDocument14 pagesSi-COAT® 579-SDSSarvenaz PakianNo ratings yet
- 2-Corrosion Rate MeasurementDocument32 pages2-Corrosion Rate Measurement이선엽100% (1)
- Nickel Free High Nitrogen Austenitic Steels - 1996 - ISIJ InternationalDocument9 pagesNickel Free High Nitrogen Austenitic Steels - 1996 - ISIJ Internationalsmith willNo ratings yet
- Passive Adsorption of Proteins: Document Version 1.1Document3 pagesPassive Adsorption of Proteins: Document Version 1.1jokonudiNo ratings yet
- Comprehensive Understanding of Grinding Aids - SikaDocument12 pagesComprehensive Understanding of Grinding Aids - SikaNam HuynhNo ratings yet
- Problems in Packing The ColumnDocument14 pagesProblems in Packing The ColumnNashia Farid0% (1)
- Basic Principles: of Non-Contact Temperature MeasurementDocument32 pagesBasic Principles: of Non-Contact Temperature Measurementchitturi jagadishNo ratings yet
- TDS en - Cip 100Document2 pagesTDS en - Cip 100CiroNo ratings yet
- Classification of Dosage FormsDocument25 pagesClassification of Dosage FormsNimit Jain100% (1)
- Experimentation of Composite Repair Techniques For PipelinesDocument13 pagesExperimentation of Composite Repair Techniques For Pipelinesusto2014No ratings yet
- The Effectiveness of Cassava Starch As A Component of Biodegradable PlasticDocument43 pagesThe Effectiveness of Cassava Starch As A Component of Biodegradable PlasticRhea Danica AngusNo ratings yet



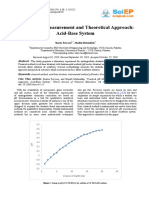





















































































Download as pdf or txt
You might also like
- Advanced Acids and BasesDocument13 pagesAdvanced Acids and BasesJohn Carlo MacalagayNo ratings yet
- Exercise 1 and 2 - Revised2Document5 pagesExercise 1 and 2 - Revised2Vivekka Olivia JohnNo ratings yet
- 4 Heat Exchanger 2Document42 pages4 Heat Exchanger 2januari desemberNo ratings yet
- WjceDocument5 pagesWjceshmohiuddinNo ratings yet
- Flatt Ve ArkDocument6 pagesFlatt Ve ArkMert GüngörNo ratings yet
- GaslawconstantDocument6 pagesGaslawconstantSefa Ceren KANDEMİRNo ratings yet
- Determination of The Molecular Weight of A Non-Volatile Solid by The Cryoscopic MethodDocument13 pagesDetermination of The Molecular Weight of A Non-Volatile Solid by The Cryoscopic Methodariana religiosoNo ratings yet
- Steam Z FactorDocument7 pagesSteam Z Factorralf.stein9265No ratings yet
- Ie13 2 0170Document6 pagesIe13 2 0170jnwxNo ratings yet
- Ubc 1999-464040Document235 pagesUbc 1999-464040Pavita SalsabilaNo ratings yet
- Assessment of Calculation Methods For Calcium Carbonate Saturation in Drinking Water For DIN 38404-10 ComplianceDocument10 pagesAssessment of Calculation Methods For Calcium Carbonate Saturation in Drinking Water For DIN 38404-10 ComplianceinejattNo ratings yet
- Calculation of Solvation Free Energies of Charged Solutes Using Mixed Cluster/Continuum ModelsDocument11 pagesCalculation of Solvation Free Energies of Charged Solutes Using Mixed Cluster/Continuum ModelsMohon GuptaNo ratings yet
- Ba Doped Co POMDocument7 pagesBa Doped Co POMpandiaraj1988No ratings yet
- Calculation of Phase Diagrams of Gas-HydratesDocument9 pagesCalculation of Phase Diagrams of Gas-HydratesMichael ParkerNo ratings yet
- Uyfrbfhrb 2Document19 pagesUyfrbfhrb 2Bill644No ratings yet
- Hay Duk 1982Document5 pagesHay Duk 1982FSBollNo ratings yet
- RyznarDocument12 pagesRyznarJim FrenkenNo ratings yet
- X 2 G 63 TSANDOVALDocument13 pagesX 2 G 63 TSANDOVALChemical Engineering Dance CrewNo ratings yet
- Sifat KoligatifDocument12 pagesSifat Koligatiflilik supriantiNo ratings yet
- Characterization of Partially Hydrolyzed Poly (Vinyl Alcohol) - Effect of Poly (Vinyl Alcohol) Molecular Architecture On Aqueous Phase ConformationDocument8 pagesCharacterization of Partially Hydrolyzed Poly (Vinyl Alcohol) - Effect of Poly (Vinyl Alcohol) Molecular Architecture On Aqueous Phase ConformationIrvan YudhistiraNo ratings yet
- Water Index CalculationsDocument24 pagesWater Index CalculationsGustavo Adolfo Piñero Borges100% (1)
- Humberto Hugo Soriano ValdiviaDocument12 pagesHumberto Hugo Soriano ValdiviaBeto SorianoNo ratings yet
- CangrumaDocument10 pagesCangrumaAna ChisNo ratings yet
- 6211 METHANEDocument3 pages6211 METHANERuth Coronado ChuyesNo ratings yet
- Ilcpa 5 20Document15 pagesIlcpa 5 20widya nurul ainiNo ratings yet
- Ion Exchange Report - Group 4 Section 2Document18 pagesIon Exchange Report - Group 4 Section 2razan.tarabay.26No ratings yet
- 1 s2.0 S1876610211005960 MainDocument8 pages1 s2.0 S1876610211005960 MainRenalyn TorioNo ratings yet
- Prediction of Scale Problems Due To Injection of Incompatible WatersDocument12 pagesPrediction of Scale Problems Due To Injection of Incompatible WatersMiguel GrNo ratings yet
- Uyfrbfhrb 3Document18 pagesUyfrbfhrb 3Bill644No ratings yet
- Cheng 2013Document10 pagesCheng 2013Cristian CastilloNo ratings yet
- A Compositional Model For CO2 Floods Including CO2 Solubility in WaterDocument13 pagesA Compositional Model For CO2 Floods Including CO2 Solubility in Watermoji20067147No ratings yet
- Chapter7 PDFDocument36 pagesChapter7 PDFnillnjtNo ratings yet
- Practica ConductimetriaDocument21 pagesPractica Conductimetriajoss villagomezNo ratings yet
- Bord PaperkorrDocument9 pagesBord Paperkorrjakalae5263No ratings yet
- EXP2 Molar Mass of A Volatile Liquid Final ReportDocument14 pagesEXP2 Molar Mass of A Volatile Liquid Final ReportBINSAHNo ratings yet
- 10.1021 Je034179fDocument5 pages10.1021 Je034179faminbm.pt24No ratings yet
- Paper No.: Corrosion Behavior of Carbon Steel in Supercritical Co - Water EnvironmentsDocument20 pagesPaper No.: Corrosion Behavior of Carbon Steel in Supercritical Co - Water Environmentss arvelakisNo ratings yet
- رێلوەه-نیددەحلاەس یۆکناز Salahaddin University-Erbil: (solubility product constant)Document8 pagesرێلوەه-نیددەحلاەس یۆکناز Salahaddin University-Erbil: (solubility product constant)shko noshaNo ratings yet
- Jurnal 5 - (Cox Et Al., 2015 - Henry Constant ReferenceDocument13 pagesJurnal 5 - (Cox Et Al., 2015 - Henry Constant Referenceanik suciNo ratings yet
- Catalysts: New Method of Determining Kinetic Parameters For Decomposition of Hydrogen Peroxide by CatalaseDocument12 pagesCatalysts: New Method of Determining Kinetic Parameters For Decomposition of Hydrogen Peroxide by CatalaseSo PhiaNo ratings yet
- IP 3. Protocol - Chemical Principles II LaboratoryDocument9 pagesIP 3. Protocol - Chemical Principles II LaboratoryJavier PratdesabaNo ratings yet
- Chemistry LaboratoryDocument31 pagesChemistry LaboratoryBunty KhiljiNo ratings yet
- Softening: Drinking Water - Lab ExperimentsDocument6 pagesSoftening: Drinking Water - Lab Experimentsnermeen ahmedNo ratings yet
- Kinetics of Iodide-Catalyzed Decomposition of Hydrogen Peroxide in A Continuous-Stirred Tank ReactorDocument6 pagesKinetics of Iodide-Catalyzed Decomposition of Hydrogen Peroxide in A Continuous-Stirred Tank ReactorGlen TrinidadNo ratings yet
- Reservoir Simulation ModelDocument6 pagesReservoir Simulation ModelDeejayF1No ratings yet
- Solubility Measurement and Modeling For PropaneDocument8 pagesSolubility Measurement and Modeling For PropanerschirtNo ratings yet
- Datos IsopiésticoDocument4 pagesDatos IsopiésticoJuanMeMooMillaNo ratings yet
- Improving The Hydrogen Oxidation Reaction Rate by Promotion of Hydroxyl AdsorptionDocument7 pagesImproving The Hydrogen Oxidation Reaction Rate by Promotion of Hydroxyl AdsorptionHasanulKamilNo ratings yet
- 0437 0454 PDFDocument18 pages0437 0454 PDFzhyhhNo ratings yet
- 0437 0454 PDFDocument18 pages0437 0454 PDFzhyhhNo ratings yet
- Thermodynamic Models For Determination of The Solubility of F2CDocument6 pagesThermodynamic Models For Determination of The Solubility of F2Ctatchanok1525No ratings yet
- Glycol Dehydration of High-Acid Gas StreamsDocument8 pagesGlycol Dehydration of High-Acid Gas StreamsAqsam NaveedNo ratings yet
- Sample Chemistry Undergraduate Laboratory ReportDocument14 pagesSample Chemistry Undergraduate Laboratory ReportApril TapayanNo ratings yet
- 1 s2.0 S001346862101999X MainDocument13 pages1 s2.0 S001346862101999X MainROBSON CardosoNo ratings yet
- Energies 17 02356 v2Document39 pagesEnergies 17 02356 v2Niraj ThakreNo ratings yet
- PH Meter It's Applications: Practical Advance Pharmaceutical AnalysisDocument19 pagesPH Meter It's Applications: Practical Advance Pharmaceutical AnalysisarjunkumbharchemNo ratings yet
- 00071326Document12 pages00071326theoneandonly1No ratings yet
- Caoh2 3rd EdDocument9 pagesCaoh2 3rd Edcano96No ratings yet
- SPE-203762-MS Using Difference Factor Thermodynamics Models in Hydrate Formation Prediction During Surface Production Well TestingDocument14 pagesSPE-203762-MS Using Difference Factor Thermodynamics Models in Hydrate Formation Prediction During Surface Production Well TestingTheNourEldenNo ratings yet
- Water Rock InteractionDocument4 pagesWater Rock InteractionPritam RajNo ratings yet
- Zhang 2014Document7 pagesZhang 2014Jordy Romario Grados SoriaNo ratings yet
- Steam TracingDocument18 pagesSteam TracingSyed Mujtaba Ali Bukhari100% (2)
- Us 5221800Document7 pagesUs 5221800Mochamad Abdul MalikNo ratings yet
- Colorkote: Technical Data SheetDocument2 pagesColorkote: Technical Data Sheet김종태No ratings yet
- TEX - CHEM 103 Organic ChemistryDocument52 pagesTEX - CHEM 103 Organic ChemistrychioNo ratings yet
- Perfringens Agar Base (Tryptose Sulphite Cycloserine Agar Base) (Iso 7937 2004, Iso 14189 2013) - TD-TM 1826Document3 pagesPerfringens Agar Base (Tryptose Sulphite Cycloserine Agar Base) (Iso 7937 2004, Iso 14189 2013) - TD-TM 1826Ventas LabsupplyNo ratings yet
- Prob11 25Document1 pageProb11 25ozi125_2No ratings yet
- EN-GJS-450-10, ASTM A536 65-45-12, Ductile Iron, SG Iron Chemical Composition, Mechanical and Physical PropertiesDocument2 pagesEN-GJS-450-10, ASTM A536 65-45-12, Ductile Iron, SG Iron Chemical Composition, Mechanical and Physical PropertiesBbiettg AndiarNo ratings yet
- Ficha Tecnica Compresor FiacDocument10 pagesFicha Tecnica Compresor FiacSAUL OSPINONo ratings yet
- Petroleum Exploration MetDocument10 pagesPetroleum Exploration MetShiraz NajatNo ratings yet
- E7010 P1 - 15680891Document4 pagesE7010 P1 - 15680891Arvind SahaniNo ratings yet
- Biosurfactant 7Document8 pagesBiosurfactant 7Aranrie MosesNo ratings yet
- Selection Control ValvesDocument7 pagesSelection Control ValvesVeera Mani100% (1)
- EXPLOSIVE CLASSIFICATIONS ATexDocument5 pagesEXPLOSIVE CLASSIFICATIONS ATexJeet SinghNo ratings yet
- Ez-10 MSDSDocument5 pagesEz-10 MSDSMark Evan SalutinNo ratings yet
- Syltherm 800 Silicone Heat Transfer Fluid MSDSDocument8 pagesSyltherm 800 Silicone Heat Transfer Fluid MSDSNasser AlshalwyNo ratings yet
- Acs Jchemed 7b00739 PDFDocument9 pagesAcs Jchemed 7b00739 PDFDaniel Ernesto Ruiz MondragonNo ratings yet
- Reflex Plus: The Powder Solve ApproachDocument4 pagesReflex Plus: The Powder Solve Approachmetal2567No ratings yet
- Si-COAT® 579-SDSDocument14 pagesSi-COAT® 579-SDSSarvenaz PakianNo ratings yet
- 2-Corrosion Rate MeasurementDocument32 pages2-Corrosion Rate Measurement이선엽100% (1)
- Nickel Free High Nitrogen Austenitic Steels - 1996 - ISIJ InternationalDocument9 pagesNickel Free High Nitrogen Austenitic Steels - 1996 - ISIJ Internationalsmith willNo ratings yet
- Passive Adsorption of Proteins: Document Version 1.1Document3 pagesPassive Adsorption of Proteins: Document Version 1.1jokonudiNo ratings yet
- Comprehensive Understanding of Grinding Aids - SikaDocument12 pagesComprehensive Understanding of Grinding Aids - SikaNam HuynhNo ratings yet
- Problems in Packing The ColumnDocument14 pagesProblems in Packing The ColumnNashia Farid0% (1)
- Basic Principles: of Non-Contact Temperature MeasurementDocument32 pagesBasic Principles: of Non-Contact Temperature Measurementchitturi jagadishNo ratings yet
- TDS en - Cip 100Document2 pagesTDS en - Cip 100CiroNo ratings yet
- Classification of Dosage FormsDocument25 pagesClassification of Dosage FormsNimit Jain100% (1)
- Experimentation of Composite Repair Techniques For PipelinesDocument13 pagesExperimentation of Composite Repair Techniques For Pipelinesusto2014No ratings yet
- The Effectiveness of Cassava Starch As A Component of Biodegradable PlasticDocument43 pagesThe Effectiveness of Cassava Starch As A Component of Biodegradable PlasticRhea Danica AngusNo ratings yet