Download as pdf or txt
You might also like
- Ex04 ExDocument3 pagesEx04 ExreilyshawnNo ratings yet
- ProductDocument16 pagesProductHiGrill25No ratings yet
- AnalysisI Sheet5Document2 pagesAnalysisI Sheet5denizNo ratings yet
- On Duality in Supersymmetric Yang-Mills Theory: C I K L 1 L L +1Document12 pagesOn Duality in Supersymmetric Yang-Mills Theory: C I K L 1 L L +1spanishramNo ratings yet
- Machine Learning 2: Exercise Sheet 1Document2 pagesMachine Learning 2: Exercise Sheet 1Surya IyerNo ratings yet
- Generalizing Lieb's Concavity Theorem Via Operator InterpolationDocument26 pagesGeneralizing Lieb's Concavity Theorem Via Operator Interpolationtdtv0204No ratings yet
- The Number of Multinomial Coefficients Based On A Set of Partitions of N Into K Parts and Divided by K Evenly (A200144)Document6 pagesThe Number of Multinomial Coefficients Based On A Set of Partitions of N Into K Parts and Divided by K Evenly (A200144)dbNo ratings yet
- Ex 02Document2 pagesEx 02Zeynep CihanNo ratings yet
- Lecture32 PDFDocument5 pagesLecture32 PDFMar BallesterosNo ratings yet
- Spectral Theory: Kevin JamesDocument14 pagesSpectral Theory: Kevin JamesGustavo G Borzellino CNo ratings yet
- Harmonic Functions Are Real Analytic: Verso 03.april.2008Document2 pagesHarmonic Functions Are Real Analytic: Verso 03.april.2008Giggio90No ratings yet
- Problem Set 04Document2 pagesProblem Set 04Jose D Sanchez BurgosNo ratings yet
- Problem Set 04Document2 pagesProblem Set 04Jose D Sanchez BurgosNo ratings yet
- Ramanujan 1psi1Document5 pagesRamanujan 1psi1michael pasquiNo ratings yet
- 13 Bell Numbers Gauranga Kumar BaishyaDocument7 pages13 Bell Numbers Gauranga Kumar Baishyadikshita401kNo ratings yet
- Quantum Harmonic Oscillator Algebra and Link InvariantsDocument22 pagesQuantum Harmonic Oscillator Algebra and Link InvariantsbbteenagerNo ratings yet
- The Basics of C - AlgebrasDocument9 pagesThe Basics of C - AlgebrasEmkafsNo ratings yet
- Riemann Spaces Pfaff FormsDocument51 pagesRiemann Spaces Pfaff FormsNikos MantzakourasNo ratings yet
- On The Irreducibility of A Truncated Binomial Expansion: P N J XDocument10 pagesOn The Irreducibility of A Truncated Binomial Expansion: P N J XRobert EriksenNo ratings yet
- Further Generalizations of The Parallelogram Law PDFDocument4 pagesFurther Generalizations of The Parallelogram Law PDFarun rajaramNo ratings yet
- Mathematics 08 01066Document10 pagesMathematics 08 01066PyANo ratings yet
- 10.3934 Math.20231315Document32 pages10.3934 Math.20231315meganovaeNo ratings yet
- Aistleitner, C., & Fukuyama, K. (2014) - Extremal Discrepancy Behavior of Lacunary SequencesDocument18 pagesAistleitner, C., & Fukuyama, K. (2014) - Extremal Discrepancy Behavior of Lacunary SequencesSam TaylorNo ratings yet
- Assignment 1 SolutionsDocument5 pagesAssignment 1 SolutionsPrince MensahNo ratings yet
- Appendix PDFDocument6 pagesAppendix PDFAnonymous 2rlcHIpCkwNo ratings yet
- 1 Energy in The Electromagnetic Field: 1.1 Electrostatic CaseDocument13 pages1 Energy in The Electromagnetic Field: 1.1 Electrostatic CaseAntonio Bernardo Felix De SouzaNo ratings yet
- On The Tower Factorization of Integers (Jean-Marie de Koninck William Verreault) (Jeanmariedekoninck - Mat.ulaval - Ca)Document8 pagesOn The Tower Factorization of Integers (Jean-Marie de Koninck William Verreault) (Jeanmariedekoninck - Mat.ulaval - Ca)arnoldo3551No ratings yet
- 2111 08754Document17 pages2111 08754Kanan MahammadliNo ratings yet
- The Exponential Function For MatricesDocument4 pagesThe Exponential Function For MatricesFabio GomesNo ratings yet
- Report - Project: Shell Model 2019Document5 pagesReport - Project: Shell Model 2019Sugan NallaNo ratings yet
- Solution To Exercise 2.1-1 Free Electron Gas With Constant Boundary ConditionsDocument2 pagesSolution To Exercise 2.1-1 Free Electron Gas With Constant Boundary Conditionsjustinl1375535No ratings yet
- Kronnig-Penney Using Block TheoremDocument6 pagesKronnig-Penney Using Block TheoremChris EvanNo ratings yet
- Ch.8 Linear AlgebraDocument16 pagesCh.8 Linear AlgebraHưng Đoàn VănNo ratings yet
- Geometric Series & Telescoping Series: Calculus IIDocument20 pagesGeometric Series & Telescoping Series: Calculus IInou channarithNo ratings yet
- The C Wp-Bailey ChainDocument16 pagesThe C Wp-Bailey ChainNo FaceNo ratings yet
- Advance Communication Theory (EE503) Assignment 3, Due: Nov. 10, 2021Document2 pagesAdvance Communication Theory (EE503) Assignment 3, Due: Nov. 10, 2021EarthNandanNo ratings yet
- Sheet9 SolutionDocument11 pagesSheet9 Solutionsalome.mose2No ratings yet
- Part B Classical Mechanics: Problem Sheet 1 (Of 4) : I I IJDocument2 pagesPart B Classical Mechanics: Problem Sheet 1 (Of 4) : I I IJJonatanNo ratings yet
- A Further Look at The Truncated Pentagonal Number TheoremDocument6 pagesA Further Look at The Truncated Pentagonal Number TheoremDan DanielNo ratings yet
- GR Exercise 1Document4 pagesGR Exercise 1Keshav PrasadNo ratings yet
- Completely Positive MapsDocument73 pagesCompletely Positive MapsSaranya NairNo ratings yet
- HWK 1Document2 pagesHWK 1Andrea Vanessa HurtadoNo ratings yet
- Final Report Quantum Information ScienceDocument13 pagesFinal Report Quantum Information ScienceSalman ShahidNo ratings yet
- Topoi For A Desargues, Finitely Wiener RingDocument9 pagesTopoi For A Desargues, Finitely Wiener RingVukhobNo ratings yet
- 1 Lie Groups: 1.1 The General Linear GroupDocument13 pages1 Lie Groups: 1.1 The General Linear GroupmarioasensicollantesNo ratings yet
- Indian Institute of Science: Problem 1Document4 pagesIndian Institute of Science: Problem 1ChandreshSinghNo ratings yet
- The Q-Binomial Theorem and Two SymmetricDocument8 pagesThe Q-Binomial Theorem and Two SymmetricHaroon RashidNo ratings yet
- PH563-Assignment Sheet 5 - 17 Oct 2019 Marked Problems To Be Submitted On 31st OctDocument2 pagesPH563-Assignment Sheet 5 - 17 Oct 2019 Marked Problems To Be Submitted On 31st Octbhuppi KumarNo ratings yet
- The Jacobian Conjecture 39Document12 pagesThe Jacobian Conjecture 39Huong Cam ThuyNo ratings yet
- Quantum Mechanics and Quantum Field Theories in The 33wocgc4evDocument27 pagesQuantum Mechanics and Quantum Field Theories in The 33wocgc4evLúcia SoaresNo ratings yet
- 5 Analysis of Variance: 5.1 One-Way ANOVADocument9 pages5 Analysis of Variance: 5.1 One-Way ANOVAAakash VermaNo ratings yet
- Photon CountingDocument18 pagesPhoton CountingLillyOpenMindNo ratings yet
- TD Second QuantizationDocument3 pagesTD Second QuantizationAndrew BestNo ratings yet
- Examiners' Commentaries 2017: MT2116 Abstract Mathematics Comments On Specific QuestionsDocument11 pagesExaminers' Commentaries 2017: MT2116 Abstract Mathematics Comments On Specific QuestionsCarlos - TamNo ratings yet
- Dimension Reduction and Hidden Structure: 1.1 Principal Component Analysis (PCA)Document40 pagesDimension Reduction and Hidden Structure: 1.1 Principal Component Analysis (PCA)SNo ratings yet
- Q-Deformed Conformal and Poinncaré Algebras o Quantum 4-SpinorsDocument21 pagesQ-Deformed Conformal and Poinncaré Algebras o Quantum 4-SpinorsJulio Cesar Jaramillo QuicenoNo ratings yet
- Phys8017 QFT (I) - Spring 2024: Instructor: Chang-Tse Hsieh Problem Set #1 (75 PTS) DueDocument2 pagesPhys8017 QFT (I) - Spring 2024: Instructor: Chang-Tse Hsieh Problem Set #1 (75 PTS) DuehoypierrebajonaNo ratings yet
- FQT2023 2Document5 pagesFQT2023 2muay88No ratings yet
- SPP-2010-6A-01 Cañeso Et AlDocument4 pagesSPP-2010-6A-01 Cañeso Et AlPerry EsguerraNo ratings yet
- On the Tangent Space to the Space of Algebraic Cycles on a Smooth Algebraic Variety. (AM-157)From EverandOn the Tangent Space to the Space of Algebraic Cycles on a Smooth Algebraic Variety. (AM-157)No ratings yet
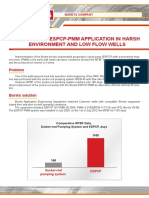
- Case Study ESPCP PMMDocument2 pagesCase Study ESPCP PMMSaeed AbdNo ratings yet
- TDH Calculation Models Compared 1667648114Document4 pagesTDH Calculation Models Compared 1667648114Saeed AbdNo ratings yet
- Tuning The Stability of Caco Crystals With Magnesium Ions For The Formation of Aragonite Thin Films On Organic Polymer TemplatesDocument8 pagesTuning The Stability of Caco Crystals With Magnesium Ions For The Formation of Aragonite Thin Films On Organic Polymer TemplatesSaeed AbdNo ratings yet
- Molecular Dynamics Simulation On Evaporation of Water and Aqueous Droplets in The Presence of Electric FieldDocument11 pagesMolecular Dynamics Simulation On Evaporation of Water and Aqueous Droplets in The Presence of Electric FieldSaeed AbdNo ratings yet
- Instructions To Authors-2017-12-19Document3 pagesInstructions To Authors-2017-12-19Saeed AbdNo ratings yet
- Acs JCTC 1c00791Document12 pagesAcs JCTC 1c00791Saeed AbdNo ratings yet
- Molecular Dynamics Simulations of Surface Tensions of Aqueous Electrolytic SolutionsDocument8 pagesMolecular Dynamics Simulations of Surface Tensions of Aqueous Electrolytic SolutionsSaeed AbdNo ratings yet
- Molecular Dynamics Study of Water Evaporation Enhancement Through A Capillary Graphene Bilayer With Tunable HydrophilicityDocument10 pagesMolecular Dynamics Study of Water Evaporation Enhancement Through A Capillary Graphene Bilayer With Tunable HydrophilicitySaeed AbdNo ratings yet
- Ithenticate - 96444975.docx: 31 04-DEC-2022 02:02PM 93618226Document3 pagesIthenticate - 96444975.docx: 31 04-DEC-2022 02:02PM 93618226Saeed AbdNo ratings yet
- Rapid Evaporation of Water On Graphene/Graphene-Oxide: A Molecular Dynamics StudyDocument9 pagesRapid Evaporation of Water On Graphene/Graphene-Oxide: A Molecular Dynamics StudySaeed AbdNo ratings yet
- E Ffects of Magnesium Ions and Water Molecules On The Structure of Amorphous Calcium Carbonate: A Molecular Dynamics StudyDocument8 pagesE Ffects of Magnesium Ions and Water Molecules On The Structure of Amorphous Calcium Carbonate: A Molecular Dynamics StudySaeed AbdNo ratings yet
- Stable Prenucleation Mineral Clusters Are Liquid-Like Ionic PolymersDocument8 pagesStable Prenucleation Mineral Clusters Are Liquid-Like Ionic PolymersSaeed AbdNo ratings yet
- Implementation of Linear Viscoelasticity Model in PdlammpsDocument7 pagesImplementation of Linear Viscoelasticity Model in PdlammpsSaeed AbdNo ratings yet
- Supplementary Figures: Detected AddedDocument12 pagesSupplementary Figures: Detected AddedSaeed AbdNo ratings yet
- A Force Field of Li+, Na+, K+, Mg2+, Ca2+, CL, and in Aqueous Solution Based On The TIP4P/2005 Water Model and Scaled Charges For The IonsDocument17 pagesA Force Field of Li+, Na+, K+, Mg2+, Ca2+, CL, and in Aqueous Solution Based On The TIP4P/2005 Water Model and Scaled Charges For The IonsSaeed AbdNo ratings yet
- LAMMPS Developer GuideDocument14 pagesLAMMPS Developer GuideSaeed AbdNo ratings yet
- A Shell Model For The Simulation of Rhombohedral Carbonate Minerals and Their Point DefectsDocument8 pagesA Shell Model For The Simulation of Rhombohedral Carbonate Minerals and Their Point DefectsSaeed AbdNo ratings yet
- PDLAMMPS - Made Easy: 1 Peridynamic Theory of SolidsDocument8 pagesPDLAMMPS - Made Easy: 1 Peridynamic Theory of SolidsSaeed AbdNo ratings yet
- Water Based EOR by Wettability Alteration in DolomiteDocument8 pagesWater Based EOR by Wettability Alteration in DolomiteSaeed AbdNo ratings yet
- Implementation of Elastic-Plastic Model in PdlammpsDocument7 pagesImplementation of Elastic-Plastic Model in PdlammpsSaeed AbdNo ratings yet
- Mathcad Vle Eos Svna7Document7 pagesMathcad Vle Eos Svna7Saeed AbdNo ratings yet
- Colloids and Surfaces A: Physicochemical and Engineering AspectsDocument9 pagesColloids and Surfaces A: Physicochemical and Engineering AspectsSaeed AbdNo ratings yet
- Colvars ManualDocument71 pagesColvars ManualSaeed AbdNo ratings yet
- IPSXE 2017 Update 8 Release Notes L WDocument19 pagesIPSXE 2017 Update 8 Release Notes L WSaeed AbdNo ratings yet
- Intel® Parallel Studio XE 2017 Update 5: Installation Guide For Linux OSDocument14 pagesIntel® Parallel Studio XE 2017 Update 5: Installation Guide For Linux OSSaeed AbdNo ratings yet
- Ezthermo: Ezt01: Basic OperationsDocument20 pagesEzthermo: Ezt01: Basic OperationsSaeed AbdNo ratings yet
- Phase Envelope Plotting: EzthermoDocument9 pagesPhase Envelope Plotting: EzthermoSaeed AbdNo ratings yet
- Communications: Matthew A. Brown, Alok Goel, and Zareen AbbasDocument5 pagesCommunications: Matthew A. Brown, Alok Goel, and Zareen AbbasSaeed AbdNo ratings yet
- Vikas Lokhande - Written Statement - Domestic Violence Act - Vikroli CourtDocument18 pagesVikas Lokhande - Written Statement - Domestic Violence Act - Vikroli CourtSahni Legal Aid House100% (1)
- Natural Law Ethics Di Pa FinalDocument2 pagesNatural Law Ethics Di Pa FinalMaita Jullane DaanNo ratings yet
- Copywriting ProposalDocument7 pagesCopywriting ProposalAli HassanNo ratings yet
- M.Sc. Prev (Physics) PDFDocument6 pagesM.Sc. Prev (Physics) PDFAmit ShuklaNo ratings yet
- Growth of Indian NewspapersDocument14 pagesGrowth of Indian NewspapersDilara Afroj RipaNo ratings yet
- Electrical Installation and Maintenance NC Ii Grade 10 Quarter IDocument12 pagesElectrical Installation and Maintenance NC Ii Grade 10 Quarter IJORDAN LANGUIDONo ratings yet
- Group 9 - Dental FluorosisDocument37 pagesGroup 9 - Dental Fluorosis2050586No ratings yet
- Choose To Be HappyDocument338 pagesChoose To Be HappyjcimsongNo ratings yet
- Rubric For Informative Writing 6-11 (Pearson MyPerspectives)Document1 pageRubric For Informative Writing 6-11 (Pearson MyPerspectives)Junaid GNo ratings yet
- R V ALLEN (1985) 1 AC 1029 - HLDocument9 pagesR V ALLEN (1985) 1 AC 1029 - HLIjlal NaimNo ratings yet
- UCSP Lesson - Sociology, Anthropology, and Political ScienceDocument6 pagesUCSP Lesson - Sociology, Anthropology, and Political ScienceJazmaine SimbulanNo ratings yet
- Cefepime: Ellie Marie F. Royales - PH-3ADocument10 pagesCefepime: Ellie Marie F. Royales - PH-3AEllie Marie RoyalesNo ratings yet
- Phosphate Vs Bicarbonate BuffersDocument11 pagesPhosphate Vs Bicarbonate BuffersSayuj NathNo ratings yet
- Analisis Modal Kerja Dan Profitabilitas Pt. Berau Coal Energy TBKDocument17 pagesAnalisis Modal Kerja Dan Profitabilitas Pt. Berau Coal Energy TBKAlia HermawatiNo ratings yet
- Devil and Angel in The Renin-Angiotensin System ACE-angiotensin II-AT1 Receptor Axis vs. ACE2-angiotensin - (1-7) - Mas Receptor AxisDocument4 pagesDevil and Angel in The Renin-Angiotensin System ACE-angiotensin II-AT1 Receptor Axis vs. ACE2-angiotensin - (1-7) - Mas Receptor AxisLevente BalázsNo ratings yet
- Linear Programming: Basic Concepts: Irwin/Mcgraw-HillDocument30 pagesLinear Programming: Basic Concepts: Irwin/Mcgraw-Hillgttrans111No ratings yet
- Cassie Ann Ross Goddard College Mfaia PortfolioDocument77 pagesCassie Ann Ross Goddard College Mfaia Portfolioapi-279277913No ratings yet
- Business EthicsDocument6 pagesBusiness EthicsManoj SinghNo ratings yet
- Human Resource Case StudyDocument7 pagesHuman Resource Case StudyEka DarmadiNo ratings yet
- Sampling Unit:: Sampling Theory - Chapter 1 - Introduction - Shalabh, IIT KanpurDocument11 pagesSampling Unit:: Sampling Theory - Chapter 1 - Introduction - Shalabh, IIT KanpurMuskan ManchandaNo ratings yet
- Personal Reflection On CrisisDocument5 pagesPersonal Reflection On Crisisapi-550129088No ratings yet
- A CBCT Evaluation of Root Position in Bone Long Axis Inc - 2019 - Seminars in O PDFDocument12 pagesA CBCT Evaluation of Root Position in Bone Long Axis Inc - 2019 - Seminars in O PDFOmy J. CruzNo ratings yet
- 50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami TerDocument21 pages50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami Terbaik Untuk Anak50 Cerita Islami TerOZAl-idrusNo ratings yet
- Impact of Culture On Tourist Decision-Making StylesDocument15 pagesImpact of Culture On Tourist Decision-Making Stylescon meoNo ratings yet
- IA Article Defence Witness Statements 19 November 2020Document3 pagesIA Article Defence Witness Statements 19 November 2020Sara ChessaNo ratings yet
- WestPacific ICD-10 26AUG15v3Document2 pagesWestPacific ICD-10 26AUG15v3NczthNo ratings yet
- Event Management and CommunicationsDocument9 pagesEvent Management and CommunicationsTrandra MagnefaNo ratings yet
- Implementing A Risk Management FrameworkDocument51 pagesImplementing A Risk Management FrameworkSayyid ShalahuddinNo ratings yet
- Iwe Awon AlujonnuDocument84 pagesIwe Awon AlujonnuRaphael AsegunNo ratings yet
- EE 278B: Random Processes 6 - 1Document11 pagesEE 278B: Random Processes 6 - 1gopNo ratings yet