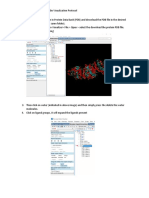
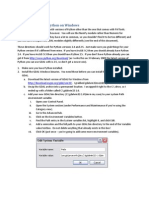
Download as pdf or txt
You might also like
- Reshade Installation Guide MSFS APRCDocument12 pagesReshade Installation Guide MSFS APRCWael ChebliNo ratings yet
- GTD Setup Guide (A4)Document41 pagesGTD Setup Guide (A4)Eden Abdula100% (7)
- Vlocity DX Setup Documentation PDFDocument2 pagesVlocity DX Setup Documentation PDFTrinesh BhargavaNo ratings yet
- ProPep 3 ManualDocument9 pagesProPep 3 Manualtony pinkNo ratings yet
- Quick Start Guide For CrystalExplorer17 - CrystalExplorer WikiDocument10 pagesQuick Start Guide For CrystalExplorer17 - CrystalExplorer WikiMarjanSplitNo ratings yet
- Vlocity DX Setup Documentation PDFDocument2 pagesVlocity DX Setup Documentation PDFTrinesh BhargavaNo ratings yet
- PyrxDocument13 pagesPyrxahsaanahmadNo ratings yet
- QsarDocument36 pagesQsarachsanuddin100% (3)
- Course Code:: PHR-322: Pharmaceutical Analysis-LlDocument7 pagesCourse Code:: PHR-322: Pharmaceutical Analysis-LlMd.Mahfuzur RahmanNo ratings yet
- Molecular Docking: in Computer Aided Drug DesignDocument26 pagesMolecular Docking: in Computer Aided Drug DesignGravelandNo ratings yet
- 13.docking ScoringDocument47 pages13.docking ScoringPranav NakhateNo ratings yet
- Protein Purification HandbookDocument94 pagesProtein Purification HandbookDolphingNo ratings yet
- Molinspiration ChembioinformaticsDocument6 pagesMolinspiration ChembioinformaticsbedibediNo ratings yet
- 2.4.20. Determination of Elemental ImpuritiesDocument4 pages2.4.20. Determination of Elemental ImpuritiessarasaNo ratings yet
- Hyperchem QSAR 2Document37 pagesHyperchem QSAR 2rafida aisyahNo ratings yet
- Exercise Z Matrix CH4Document18 pagesExercise Z Matrix CH4Manjeet BhatiaNo ratings yet
- Gibbs-Duhem Equation PDFDocument5 pagesGibbs-Duhem Equation PDFjoker0% (1)
- Quantitative Structure-Activity Relationships (QSAR)Document30 pagesQuantitative Structure-Activity Relationships (QSAR)Yoga NurwijayaNo ratings yet
- Institutional Summer Training WorksheetDocument9 pagesInstitutional Summer Training WorksheetApoorva SinghNo ratings yet
- AutoDock Tutorial PDFDocument17 pagesAutoDock Tutorial PDFrednriNo ratings yet
- DockingDocument6 pagesDockingFrancimauro MoraisNo ratings yet
- Auto Dock TutorialDocument2 pagesAuto Dock TutorialParijat DasNo ratings yet
- Anaconda InstallationDocument9 pagesAnaconda InstallationanmolNo ratings yet
- Protein Structure ExplorationDocument22 pagesProtein Structure Explorationsuveer698No ratings yet
- Illustrated Step-By-Step Protocol To Perform Molecular DockingDocument64 pagesIllustrated Step-By-Step Protocol To Perform Molecular DockingMasyitah ulfayuraNo ratings yet
- Argus Molecular Docking 1Document5 pagesArgus Molecular Docking 1escherichioNo ratings yet
- Vital Community DocumentationDocument27 pagesVital Community DocumentationEduardoNo ratings yet
- Manual BioblenderDocument28 pagesManual BioblenderFernando Henrique DeslockNo ratings yet
- Installing Pynomo On A Windows Machine: Ron Doerfler (21 December 2011)Document3 pagesInstalling Pynomo On A Windows Machine: Ron Doerfler (21 December 2011)DacianMNo ratings yet
- Autodock - Vina ProtocolDocument12 pagesAutodock - Vina ProtocolNaresh kumarNo ratings yet
- Cyber Forsenic Practical - Journal-1Document77 pagesCyber Forsenic Practical - Journal-1crazzy demonNo ratings yet
- ( ( ( ( Questions Which Are Not Taken From GeekinterviewDocument4 pages( ( ( ( Questions Which Are Not Taken From GeekinterviewAbhilasha ModekarNo ratings yet
- How To Write Assembly Programs in Keil: Sepehr NaimiDocument11 pagesHow To Write Assembly Programs in Keil: Sepehr Naimiraghavendran_beeeeNo ratings yet
- Re Cross Docking Tutorial 27aug12Document5 pagesRe Cross Docking Tutorial 27aug12dulNo ratings yet
- Gdal WinDocument3 pagesGdal WinJorge Enrique Suárez CardosoNo ratings yet
- Tutorial Docking With GlideDocument5 pagesTutorial Docking With GlideJohn M. E. PurbaNo ratings yet
- Verification Guide - Results Display and Forum PublishingDocument8 pagesVerification Guide - Results Display and Forum PublishingMuhamed HalilovicNo ratings yet
- Hazardous Material Ordering Reference Guide (Ver 1) PDFDocument53 pagesHazardous Material Ordering Reference Guide (Ver 1) PDFLj PerrierNo ratings yet
- Description: Tags: PISA2003CB QuickguideDocument34 pagesDescription: Tags: PISA2003CB Quickguideanon-266064No ratings yet
- Installation of IBM Cognos 8 - 4 - by HarishDocument9 pagesInstallation of IBM Cognos 8 - 4 - by HarishsharpanNo ratings yet
- Geoserver ManualDocument42 pagesGeoserver ManualTomi Toivio100% (2)
- TG110-Dtpw Manual 207 With USB AddendumDocument34 pagesTG110-Dtpw Manual 207 With USB Addendumigniz16No ratings yet
- Seanmaro 04 Alignment-WorkshopDocument26 pagesSeanmaro 04 Alignment-Workshopfda.narvaezNo ratings yet
- Harmonising Bibliographic Data Using OpenRefineDocument5 pagesHarmonising Bibliographic Data Using OpenRefineraum123No ratings yet
- Peer Guardian User Guide enDocument22 pagesPeer Guardian User Guide engeekweekNo ratings yet
- Exp 6Document12 pagesExp 6Soban MarufNo ratings yet
- Updated OpenFAST User Manual Windows 10Document51 pagesUpdated OpenFAST User Manual Windows 10Mahdi MahnazNo ratings yet
- AUDocker LE ManualDocument11 pagesAUDocker LE ManualdehammoNo ratings yet
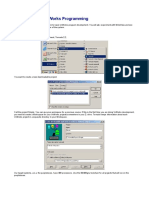
- Introduction To Vxworks Programming: Start A Vxworks ProjectDocument11 pagesIntroduction To Vxworks Programming: Start A Vxworks ProjectJarot SugihartoNo ratings yet
- RobotLoad User Reference 2Document10 pagesRobotLoad User Reference 2Vinodh VijayakumarNo ratings yet
- Toad Installation GuideDocument4 pagesToad Installation GuideRex PeñarandaNo ratings yet
- IP User - S Guide BorjaDocument32 pagesIP User - S Guide BorjaFranco Palenque ValdezNo ratings yet
- Chemsep Tutorial: Pcdmanager: Ross Taylor and Harry Kooijman Updated For Version 6.8, August 1, 2011Document37 pagesChemsep Tutorial: Pcdmanager: Ross Taylor and Harry Kooijman Updated For Version 6.8, August 1, 2011joseNo ratings yet
- 0 Anaconda-Guide 040323Document22 pages0 Anaconda-Guide 040323Ahmad IlhamNo ratings yet
- Create Exe of Java ProjectDocument12 pagesCreate Exe of Java Projectdewang goelNo ratings yet
- Windows Installation Guide For Suricata IDS/IPS/NSMDocument41 pagesWindows Installation Guide For Suricata IDS/IPS/NSMEDUARDONo ratings yet
- Python - Installation Instruction - WindowsDocument101 pagesPython - Installation Instruction - WindowsClaudiu StefanNo ratings yet
- Lab 01 - Getting - StartedDocument13 pagesLab 01 - Getting - StartedMuath AfeshatNo ratings yet
- 9000XDrive User ManualDocument28 pages9000XDrive User ManualagroelizabethNo ratings yet
- Introduction To HYSYS Steady StateDocument22 pagesIntroduction To HYSYS Steady StatebehnamhfNo ratings yet
- Furcadia Installer Browser (FIB)Document9 pagesFurcadia Installer Browser (FIB)Artex // IceDragonNo ratings yet
- كتاب الطواسين لأبي المغيث الحسين بن منصور الحلاجDocument46 pagesكتاب الطواسين لأبي المغيث الحسين بن منصور الحلاجMohamed Reda86% (7)
- Mattepally Vishwaja: ProjectDocument3 pagesMattepally Vishwaja: ProjectVishwaja MattepallyNo ratings yet
- Axp202 X PowersDocument49 pagesAxp202 X PowersromenoNo ratings yet
- Sap Is UDocument3 pagesSap Is Uyarrapradeep0% (1)
- Git GithubDocument21 pagesGit GithubKeshav goyalNo ratings yet
- Flexible Automation: Z Z Z ZDocument12 pagesFlexible Automation: Z Z Z ZMuhammad HafizNo ratings yet
- Vicinity Map: Ralph Luis D. Cardano Adamson University Ian Ray L. DecenaDocument1 pageVicinity Map: Ralph Luis D. Cardano Adamson University Ian Ray L. DecenaGecilo TabogonNo ratings yet
- An Industrial Training ReportDocument43 pagesAn Industrial Training ReportRutuja PoteNo ratings yet
- Web Based Programming 1 Syllabus TemplateDocument5 pagesWeb Based Programming 1 Syllabus TemplateHanz Dela CruzNo ratings yet
- P120 OrderForm - V13 - 052020Document13 pagesP120 OrderForm - V13 - 052020haroldNo ratings yet
- Philips Ds9800w-10!37!93 Ver.1.1 Docking Speaker SystemDocument36 pagesPhilips Ds9800w-10!37!93 Ver.1.1 Docking Speaker Systemsupermax900100% (2)
- IPS Tempo: User ManualDocument262 pagesIPS Tempo: User ManualdevaseelanNo ratings yet
- EMR-3000 User Manual Eaton En2Document558 pagesEMR-3000 User Manual Eaton En2yes-caliNo ratings yet
- Microprocessor Theory and Applications With 6800068020 and Pentium TQW - DarksidergDocument590 pagesMicroprocessor Theory and Applications With 6800068020 and Pentium TQW - DarksidergSuraj Kumar Gupta100% (4)
- Interface To Network Security Functions For Cloud-BasedDocument16 pagesInterface To Network Security Functions For Cloud-BasedFauzie Arrachmad NurNo ratings yet
- The Crypto Story by Matt LevineDocument84 pagesThe Crypto Story by Matt LevinePaulo JatahyNo ratings yet
- Y7 2019.Y7 Prime 2019 PCB Layout HL3DUBMDocument2 pagesY7 2019.Y7 Prime 2019 PCB Layout HL3DUBMebrahemhsn2023100% (1)
- Marketing Project 2Document22 pagesMarketing Project 2RajatVimalNo ratings yet
- B.Tech. VII-Sem: Cad/CamDocument162 pagesB.Tech. VII-Sem: Cad/CamSushanthNo ratings yet
- Double Server Public Key Encryption With Keyword Search For Secure Cloud StorageDocument8 pagesDouble Server Public Key Encryption With Keyword Search For Secure Cloud StorageEditor IJTSRDNo ratings yet
- Seminar ReportDocument40 pagesSeminar Reportvirender24783% (6)
- Enu910gl M7 PDFDocument31 pagesEnu910gl M7 PDFkairuddinNo ratings yet
- CS-636 Compiler Construction OutlineDocument3 pagesCS-636 Compiler Construction OutlineTayyab FiazNo ratings yet
- Alcatel 4635 PDFDocument159 pagesAlcatel 4635 PDFEdmundo Melchor GarcíaNo ratings yet
- EZ-Phone (Hss Sip Ua) User's ManualDocument17 pagesEZ-Phone (Hss Sip Ua) User's ManualVenkatrama Kaushik IyerNo ratings yet
- Deploying and Managing Windows and Hyper-V ContainersDocument25 pagesDeploying and Managing Windows and Hyper-V ContainersMarculino LimaNo ratings yet
- 2 - Psion EP10 - User GuideDocument266 pages2 - Psion EP10 - User GuideEnrique LefiánNo ratings yet
- DropboxDocument3 pagesDropboxHarry Andrés Labio BNo ratings yet
- Artificial Intelligence in GamesDocument30 pagesArtificial Intelligence in GamesPhantomNo ratings yet