
Advanced Inorganic Chemistry Laboratory - Lab. Manual, Prushan
Advanced Inorganic Chemistry Laboratory - Lab. Manual, Prushan
You might also like
- Metals in MedicineDocument4 pagesMetals in MedicineSavi sharmaNo ratings yet
- Molecular Thermodynamics - Richard E. DickersonDocument470 pagesMolecular Thermodynamics - Richard E. Dickersonbohrdom100% (5)
- Spectroscopic Methods in Organic ChemistryDocument438 pagesSpectroscopic Methods in Organic ChemistryElthon Salvador Cavazos Medina100% (1)
- Advanced Potentiometry-Potentiometric Titrations and Their Systematic Errors (Erzs Ebet N Eher-Neumann-2009) PDFDocument252 pagesAdvanced Potentiometry-Potentiometric Titrations and Their Systematic Errors (Erzs Ebet N Eher-Neumann-2009) PDFNew Topics in Analytical ChemistryNo ratings yet
- Applied Homogeneous Catalysis With Organo-Metallic Compounds - 2nd EditionDocument1,492 pagesApplied Homogeneous Catalysis With Organo-Metallic Compounds - 2nd Editionharrypoutreur100% (4)
- Surface Analysis Methods in Materials ScienceDocument588 pagesSurface Analysis Methods in Materials Sciencesara pezhdad100% (2)
- Kinetics and Mehanism of Reactions of Transition Metal Complexes by Ralph G. WilkinsDocument477 pagesKinetics and Mehanism of Reactions of Transition Metal Complexes by Ralph G. WilkinsAbhishek Abhi100% (3)
- Carbonate-Bicarbonate Mixture Anal Chem Post LabDocument7 pagesCarbonate-Bicarbonate Mixture Anal Chem Post LabKennedy OrtegaNo ratings yet
- Spectroscopic Solutions of StructureDocument21 pagesSpectroscopic Solutions of StructureKassimNo ratings yet
- PMR Spectroscopy: Solved Problems Volume : IIFrom EverandPMR Spectroscopy: Solved Problems Volume : IIRating: 5 out of 5 stars5/5 (3)
- Defects and Defect Processes in Nonmetallic SolidsFrom EverandDefects and Defect Processes in Nonmetallic SolidsRating: 4 out of 5 stars4/5 (1)
- Lawson Criteria For Nuclear FusionDocument3 pagesLawson Criteria For Nuclear FusionShekhar NarwalNo ratings yet
- 3 YEAR BUILDING SERVICES-illumination PDFDocument44 pages3 YEAR BUILDING SERVICES-illumination PDFanurag gupta100% (2)
- Inorganic Lab Exp 2Document6 pagesInorganic Lab Exp 2Jekyll Rev67% (3)
- Practice Makes Perfect in Chemistry: Compounds, Reactions and MolesFrom EverandPractice Makes Perfect in Chemistry: Compounds, Reactions and MolesNo ratings yet
- Practice Makes Perfect in Chemistry: The Physical Behavior of MatterFrom EverandPractice Makes Perfect in Chemistry: The Physical Behavior of MatterRating: 5 out of 5 stars5/5 (1)
- Practice Makes Perfect in Chemistry: Compounds, Reactions and Moles with AnswersFrom EverandPractice Makes Perfect in Chemistry: Compounds, Reactions and Moles with AnswersRating: 3 out of 5 stars3/5 (2)
- IR OrganometallicDocument21 pagesIR OrganometallicYanti Yana HalidNo ratings yet
- Coordination ChemistryDocument25 pagesCoordination Chemistrypatel_346879839No ratings yet
- Metal Complexes or Coordination Compounds: Kfecn 4K Fe CNDocument90 pagesMetal Complexes or Coordination Compounds: Kfecn 4K Fe CNPavan Boro100% (1)
- Inorganic Lab1Document50 pagesInorganic Lab1Mohamed YasinNo ratings yet
- Inorganic Chemistry ExpDocument46 pagesInorganic Chemistry Exppc355chyi100% (3)
- NMR Solving StrategyDocument2 pagesNMR Solving Strategysorrow Lemon100% (1)
- Characterization of PolymersDocument41 pagesCharacterization of PolymersMomentum Press100% (1)
- Introduction of Powder Diffraction INDIADocument397 pagesIntroduction of Powder Diffraction INDIAJuan Pablo Cano Mejia100% (1)
- CH 431 Lab ManualFullDocument28 pagesCH 431 Lab ManualFullHân BảoNo ratings yet
- Organometallic Transition Metal Catalysis: A Holistic Approach to Understanding and Predicting their MechanismsFrom EverandOrganometallic Transition Metal Catalysis: A Holistic Approach to Understanding and Predicting their MechanismsNo ratings yet
- Characterization of PolymersDocument30 pagesCharacterization of Polymerssuranjana26No ratings yet
- OrganometallicsDocument58 pagesOrganometallicsRohit ChaudharyNo ratings yet
- Lucas Test PDFDocument3 pagesLucas Test PDFciciNo ratings yet
- Essentials of Pericyclic and Photo (Biswanath Dinda) PDFDocument363 pagesEssentials of Pericyclic and Photo (Biswanath Dinda) PDFKamal Kishor Thakur100% (1)
- Organic Chemistry-All Reaction MechanismsDocument30 pagesOrganic Chemistry-All Reaction MechanismsParameshwari kumar100% (1)
- Inorg Reactionn MechanismsDocument38 pagesInorg Reactionn MechanismsThabang MolekoNo ratings yet
- Ligand Field TheoryDocument27 pagesLigand Field TheoryAmir Nazri Kaibing100% (2)
- Reaction Mechanisms in Environmental Organic ChemistryDocument83 pagesReaction Mechanisms in Environmental Organic ChemistryThanh Vân Trần ThịNo ratings yet
- MGOM 1 Introduction To OrganometallicsDocument70 pagesMGOM 1 Introduction To OrganometallicsIyan Maulana100% (1)
- Chemistry of Reactive Intermediate FinalDocument38 pagesChemistry of Reactive Intermediate FinalTefera100% (1)
- Spectroscopic Properties of Inorganic and OrganicDocument375 pagesSpectroscopic Properties of Inorganic and Organicshivachem180% (1)
- Ion Exchange and Solvent Extrac 20Document365 pagesIon Exchange and Solvent Extrac 20АртемNo ratings yet
- Spectral Calculation of Orgel and Tanabe-Sugano Diagram'SDocument18 pagesSpectral Calculation of Orgel and Tanabe-Sugano Diagram'STiwari VishalNo ratings yet
- Ch1b Ps3 Key SerDocument7 pagesCh1b Ps3 Key SerRichard ZhuNo ratings yet
- EPR Spectroscopy-App in Chemistry & Biology (Topics in Current Chemistry v.321) - M.drescher & G.jeschkeDocument247 pagesEPR Spectroscopy-App in Chemistry & Biology (Topics in Current Chemistry v.321) - M.drescher & G.jeschkepplugc100% (2)
- Royal Society of Chemistry Organometallic Chemis 048 PDFDocument468 pagesRoyal Society of Chemistry Organometallic Chemis 048 PDFpi.314153.4No ratings yet
- Organometallic Chemistry: Prof DR Hadariah Bahron Organometallic Chemistry March-July 2018Document44 pagesOrganometallic Chemistry: Prof DR Hadariah Bahron Organometallic Chemistry March-July 2018Mior Afiq100% (1)
- CHAPTER 3 Phase Diagram TTT HT - 1stDocument25 pagesCHAPTER 3 Phase Diagram TTT HT - 1stAriff AziziNo ratings yet
- Report NMRDocument13 pagesReport NMRsarahNo ratings yet
- PDFDocument155 pagesPDFHifza shairwani100% (1)
- Living PolymerizationDocument58 pagesLiving PolymerizationdohuucauNo ratings yet
- Molecular RearrangementsDocument9 pagesMolecular RearrangementsDhanaswamy Ilangeswaran67% (3)
- Organic Reactions and MechanismDocument51 pagesOrganic Reactions and MechanismAbhay Kumar Nayak75% (8)
- UV Spectroscopy 2016Document87 pagesUV Spectroscopy 2016M Mudassar AslamNo ratings yet
- M.SC - Chemistry SEM 2 Syllabus 2018Document14 pagesM.SC - Chemistry SEM 2 Syllabus 2018Rukam Singh TomarNo ratings yet
- Organometallics Notes PDFDocument130 pagesOrganometallics Notes PDFBiswa Bhusan NayakNo ratings yet
- Synthetic Inorganic Chemistry Blanchard 5thed1937Document396 pagesSynthetic Inorganic Chemistry Blanchard 5thed1937rhozab100% (1)
- Metal Nitrosyl ComplexDocument6 pagesMetal Nitrosyl Complexsusanenulfah100% (2)
- M-Dinitrobenzene From Nitrobenzene by Substitution ReactionDocument22 pagesM-Dinitrobenzene From Nitrobenzene by Substitution ReactionFarheen BehlimNo ratings yet
- Inverse Coordination Chemistry: A Novel Chemical Concept: Academic PrimersFrom EverandInverse Coordination Chemistry: A Novel Chemical Concept: Academic PrimersNo ratings yet
- Introduction to Voltammetric Analysis: Theory and PracticeFrom EverandIntroduction to Voltammetric Analysis: Theory and PracticeNo ratings yet
- Classification Non Verbal Reasoning Questions and AnswersDocument38 pagesClassification Non Verbal Reasoning Questions and AnswersShruti Jha100% (3)
- The Changing ManDocument22 pagesThe Changing ManMacmillan KidsNo ratings yet
- 03 Metal Complex EquilibriaDocument12 pages03 Metal Complex EquilibriaAndrew James ViernesNo ratings yet
- Polyelectrolyte Complexes in The Dispersed and Solid State I - Principles and TheoryDocument236 pagesPolyelectrolyte Complexes in The Dispersed and Solid State I - Principles and TheoryDanyelli GomesNo ratings yet
- Class02 - 08 Schematic Eyes - IntroductionDocument18 pagesClass02 - 08 Schematic Eyes - IntroductioncehborrotoNo ratings yet
- Fundamentals of Hydraulic PumpsDocument19 pagesFundamentals of Hydraulic Pumpslopez8a100% (1)
- Gandhinagar Institute of Technology: Mechanical DepartmentDocument17 pagesGandhinagar Institute of Technology: Mechanical DepartmentPAL PARTHIVNo ratings yet
- MA1001-Assg 4Document1 pageMA1001-Assg 4Tony StarkNo ratings yet
- Streamline Gud PDFDocument24 pagesStreamline Gud PDFsishu21No ratings yet
- Thomas-Fermi Screening by Lindhard FormulaDocument7 pagesThomas-Fermi Screening by Lindhard FormuladanqphyNo ratings yet
- Gas DynamicsDocument1 pageGas DynamicsBhavesh PipaliyaNo ratings yet
- Universiti Sains Malaysia School of Chemical Engineering: Pre-Lab TestDocument5 pagesUniversiti Sains Malaysia School of Chemical Engineering: Pre-Lab TestJoan MaryNo ratings yet
- Vehicle Electrical System - MATLAB & Simulink Example - MathWorks IndiaDocument2 pagesVehicle Electrical System - MATLAB & Simulink Example - MathWorks IndiaPasupuleti SivakumarNo ratings yet
- Introduction of Brownian MotionDocument10 pagesIntroduction of Brownian MotionLee Shin LeongNo ratings yet
- Cie Igcse Physics Chapter 6 2023 OnwDocument14 pagesCie Igcse Physics Chapter 6 2023 OnwZeinab ElkholyNo ratings yet
- Biot Savart LawDocument50 pagesBiot Savart LawRaveendra ReddyNo ratings yet
- Class Xii Physics Assignment 1 Unit 1Document2 pagesClass Xii Physics Assignment 1 Unit 1vishal110085No ratings yet
- Markschemes FYE Sec 3Document2 pagesMarkschemes FYE Sec 3KonicNo ratings yet
- Quantum Physics For Beginners - A Comprehensive Guide For The StarterDocument319 pagesQuantum Physics For Beginners - A Comprehensive Guide For The StarterYiannis Thomaidis100% (6)
- MTC 336 Study Guide 1228594655Document3 pagesMTC 336 Study Guide 1228594655Aaron SotoNo ratings yet
- Difference Between Static Relays and Electromagnetic RelaysDocument2 pagesDifference Between Static Relays and Electromagnetic Relayskriitka86% (7)
- 39Document26 pages39Moamen MohamedNo ratings yet
- 19 - Dowel BarsDocument22 pages19 - Dowel BarsboyzesNo ratings yet
- January 2012 MS - Unit 4 Edexcel Chemistry A-LevelDocument24 pagesJanuary 2012 MS - Unit 4 Edexcel Chemistry A-LevelMaria KolokasiNo ratings yet
- 07Document3 pages07Ruswanto Ais ZalfaNo ratings yet
- FFT Zero PadDocument4 pagesFFT Zero PadKim Gerald TejadaNo ratings yet
- Reverse OsmosisDocument24 pagesReverse OsmosisHyumi DarthNo ratings yet
- Electromotive Force Magnetic FluxDocument8 pagesElectromotive Force Magnetic FluxJalal Al-SaifNo ratings yet














































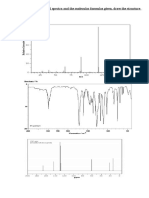










































Download as pdf or txt
You might also like
- Metals in MedicineDocument4 pagesMetals in MedicineSavi sharmaNo ratings yet
- Molecular Thermodynamics - Richard E. DickersonDocument470 pagesMolecular Thermodynamics - Richard E. Dickersonbohrdom100% (5)
- Spectroscopic Methods in Organic ChemistryDocument438 pagesSpectroscopic Methods in Organic ChemistryElthon Salvador Cavazos Medina100% (1)
- Advanced Potentiometry-Potentiometric Titrations and Their Systematic Errors (Erzs Ebet N Eher-Neumann-2009) PDFDocument252 pagesAdvanced Potentiometry-Potentiometric Titrations and Their Systematic Errors (Erzs Ebet N Eher-Neumann-2009) PDFNew Topics in Analytical ChemistryNo ratings yet
- Applied Homogeneous Catalysis With Organo-Metallic Compounds - 2nd EditionDocument1,492 pagesApplied Homogeneous Catalysis With Organo-Metallic Compounds - 2nd Editionharrypoutreur100% (4)
- Surface Analysis Methods in Materials ScienceDocument588 pagesSurface Analysis Methods in Materials Sciencesara pezhdad100% (2)
- Kinetics and Mehanism of Reactions of Transition Metal Complexes by Ralph G. WilkinsDocument477 pagesKinetics and Mehanism of Reactions of Transition Metal Complexes by Ralph G. WilkinsAbhishek Abhi100% (3)
- Carbonate-Bicarbonate Mixture Anal Chem Post LabDocument7 pagesCarbonate-Bicarbonate Mixture Anal Chem Post LabKennedy OrtegaNo ratings yet
- Spectroscopic Solutions of StructureDocument21 pagesSpectroscopic Solutions of StructureKassimNo ratings yet
- PMR Spectroscopy: Solved Problems Volume : IIFrom EverandPMR Spectroscopy: Solved Problems Volume : IIRating: 5 out of 5 stars5/5 (3)
- Defects and Defect Processes in Nonmetallic SolidsFrom EverandDefects and Defect Processes in Nonmetallic SolidsRating: 4 out of 5 stars4/5 (1)
- Lawson Criteria For Nuclear FusionDocument3 pagesLawson Criteria For Nuclear FusionShekhar NarwalNo ratings yet
- 3 YEAR BUILDING SERVICES-illumination PDFDocument44 pages3 YEAR BUILDING SERVICES-illumination PDFanurag gupta100% (2)
- Inorganic Lab Exp 2Document6 pagesInorganic Lab Exp 2Jekyll Rev67% (3)
- Practice Makes Perfect in Chemistry: Compounds, Reactions and MolesFrom EverandPractice Makes Perfect in Chemistry: Compounds, Reactions and MolesNo ratings yet
- Practice Makes Perfect in Chemistry: The Physical Behavior of MatterFrom EverandPractice Makes Perfect in Chemistry: The Physical Behavior of MatterRating: 5 out of 5 stars5/5 (1)
- Practice Makes Perfect in Chemistry: Compounds, Reactions and Moles with AnswersFrom EverandPractice Makes Perfect in Chemistry: Compounds, Reactions and Moles with AnswersRating: 3 out of 5 stars3/5 (2)
- IR OrganometallicDocument21 pagesIR OrganometallicYanti Yana HalidNo ratings yet
- Coordination ChemistryDocument25 pagesCoordination Chemistrypatel_346879839No ratings yet
- Metal Complexes or Coordination Compounds: Kfecn 4K Fe CNDocument90 pagesMetal Complexes or Coordination Compounds: Kfecn 4K Fe CNPavan Boro100% (1)
- Inorganic Lab1Document50 pagesInorganic Lab1Mohamed YasinNo ratings yet
- Inorganic Chemistry ExpDocument46 pagesInorganic Chemistry Exppc355chyi100% (3)
- NMR Solving StrategyDocument2 pagesNMR Solving Strategysorrow Lemon100% (1)
- Characterization of PolymersDocument41 pagesCharacterization of PolymersMomentum Press100% (1)
- Introduction of Powder Diffraction INDIADocument397 pagesIntroduction of Powder Diffraction INDIAJuan Pablo Cano Mejia100% (1)
- CH 431 Lab ManualFullDocument28 pagesCH 431 Lab ManualFullHân BảoNo ratings yet
- Organometallic Transition Metal Catalysis: A Holistic Approach to Understanding and Predicting their MechanismsFrom EverandOrganometallic Transition Metal Catalysis: A Holistic Approach to Understanding and Predicting their MechanismsNo ratings yet
- Characterization of PolymersDocument30 pagesCharacterization of Polymerssuranjana26No ratings yet
- OrganometallicsDocument58 pagesOrganometallicsRohit ChaudharyNo ratings yet
- Lucas Test PDFDocument3 pagesLucas Test PDFciciNo ratings yet
- Essentials of Pericyclic and Photo (Biswanath Dinda) PDFDocument363 pagesEssentials of Pericyclic and Photo (Biswanath Dinda) PDFKamal Kishor Thakur100% (1)
- Organic Chemistry-All Reaction MechanismsDocument30 pagesOrganic Chemistry-All Reaction MechanismsParameshwari kumar100% (1)
- Inorg Reactionn MechanismsDocument38 pagesInorg Reactionn MechanismsThabang MolekoNo ratings yet
- Ligand Field TheoryDocument27 pagesLigand Field TheoryAmir Nazri Kaibing100% (2)
- Reaction Mechanisms in Environmental Organic ChemistryDocument83 pagesReaction Mechanisms in Environmental Organic ChemistryThanh Vân Trần ThịNo ratings yet
- MGOM 1 Introduction To OrganometallicsDocument70 pagesMGOM 1 Introduction To OrganometallicsIyan Maulana100% (1)
- Chemistry of Reactive Intermediate FinalDocument38 pagesChemistry of Reactive Intermediate FinalTefera100% (1)
- Spectroscopic Properties of Inorganic and OrganicDocument375 pagesSpectroscopic Properties of Inorganic and Organicshivachem180% (1)
- Ion Exchange and Solvent Extrac 20Document365 pagesIon Exchange and Solvent Extrac 20АртемNo ratings yet
- Spectral Calculation of Orgel and Tanabe-Sugano Diagram'SDocument18 pagesSpectral Calculation of Orgel and Tanabe-Sugano Diagram'STiwari VishalNo ratings yet
- Ch1b Ps3 Key SerDocument7 pagesCh1b Ps3 Key SerRichard ZhuNo ratings yet
- EPR Spectroscopy-App in Chemistry & Biology (Topics in Current Chemistry v.321) - M.drescher & G.jeschkeDocument247 pagesEPR Spectroscopy-App in Chemistry & Biology (Topics in Current Chemistry v.321) - M.drescher & G.jeschkepplugc100% (2)
- Royal Society of Chemistry Organometallic Chemis 048 PDFDocument468 pagesRoyal Society of Chemistry Organometallic Chemis 048 PDFpi.314153.4No ratings yet
- Organometallic Chemistry: Prof DR Hadariah Bahron Organometallic Chemistry March-July 2018Document44 pagesOrganometallic Chemistry: Prof DR Hadariah Bahron Organometallic Chemistry March-July 2018Mior Afiq100% (1)
- CHAPTER 3 Phase Diagram TTT HT - 1stDocument25 pagesCHAPTER 3 Phase Diagram TTT HT - 1stAriff AziziNo ratings yet
- Report NMRDocument13 pagesReport NMRsarahNo ratings yet
- PDFDocument155 pagesPDFHifza shairwani100% (1)
- Living PolymerizationDocument58 pagesLiving PolymerizationdohuucauNo ratings yet
- Molecular RearrangementsDocument9 pagesMolecular RearrangementsDhanaswamy Ilangeswaran67% (3)
- Organic Reactions and MechanismDocument51 pagesOrganic Reactions and MechanismAbhay Kumar Nayak75% (8)
- UV Spectroscopy 2016Document87 pagesUV Spectroscopy 2016M Mudassar AslamNo ratings yet
- M.SC - Chemistry SEM 2 Syllabus 2018Document14 pagesM.SC - Chemistry SEM 2 Syllabus 2018Rukam Singh TomarNo ratings yet
- Organometallics Notes PDFDocument130 pagesOrganometallics Notes PDFBiswa Bhusan NayakNo ratings yet
- Synthetic Inorganic Chemistry Blanchard 5thed1937Document396 pagesSynthetic Inorganic Chemistry Blanchard 5thed1937rhozab100% (1)
- Metal Nitrosyl ComplexDocument6 pagesMetal Nitrosyl Complexsusanenulfah100% (2)
- M-Dinitrobenzene From Nitrobenzene by Substitution ReactionDocument22 pagesM-Dinitrobenzene From Nitrobenzene by Substitution ReactionFarheen BehlimNo ratings yet
- Inverse Coordination Chemistry: A Novel Chemical Concept: Academic PrimersFrom EverandInverse Coordination Chemistry: A Novel Chemical Concept: Academic PrimersNo ratings yet
- Introduction to Voltammetric Analysis: Theory and PracticeFrom EverandIntroduction to Voltammetric Analysis: Theory and PracticeNo ratings yet
- Classification Non Verbal Reasoning Questions and AnswersDocument38 pagesClassification Non Verbal Reasoning Questions and AnswersShruti Jha100% (3)
- The Changing ManDocument22 pagesThe Changing ManMacmillan KidsNo ratings yet
- 03 Metal Complex EquilibriaDocument12 pages03 Metal Complex EquilibriaAndrew James ViernesNo ratings yet
- Polyelectrolyte Complexes in The Dispersed and Solid State I - Principles and TheoryDocument236 pagesPolyelectrolyte Complexes in The Dispersed and Solid State I - Principles and TheoryDanyelli GomesNo ratings yet
- Class02 - 08 Schematic Eyes - IntroductionDocument18 pagesClass02 - 08 Schematic Eyes - IntroductioncehborrotoNo ratings yet
- Fundamentals of Hydraulic PumpsDocument19 pagesFundamentals of Hydraulic Pumpslopez8a100% (1)
- Gandhinagar Institute of Technology: Mechanical DepartmentDocument17 pagesGandhinagar Institute of Technology: Mechanical DepartmentPAL PARTHIVNo ratings yet
- MA1001-Assg 4Document1 pageMA1001-Assg 4Tony StarkNo ratings yet
- Streamline Gud PDFDocument24 pagesStreamline Gud PDFsishu21No ratings yet
- Thomas-Fermi Screening by Lindhard FormulaDocument7 pagesThomas-Fermi Screening by Lindhard FormuladanqphyNo ratings yet
- Gas DynamicsDocument1 pageGas DynamicsBhavesh PipaliyaNo ratings yet
- Universiti Sains Malaysia School of Chemical Engineering: Pre-Lab TestDocument5 pagesUniversiti Sains Malaysia School of Chemical Engineering: Pre-Lab TestJoan MaryNo ratings yet
- Vehicle Electrical System - MATLAB & Simulink Example - MathWorks IndiaDocument2 pagesVehicle Electrical System - MATLAB & Simulink Example - MathWorks IndiaPasupuleti SivakumarNo ratings yet
- Introduction of Brownian MotionDocument10 pagesIntroduction of Brownian MotionLee Shin LeongNo ratings yet
- Cie Igcse Physics Chapter 6 2023 OnwDocument14 pagesCie Igcse Physics Chapter 6 2023 OnwZeinab ElkholyNo ratings yet
- Biot Savart LawDocument50 pagesBiot Savart LawRaveendra ReddyNo ratings yet
- Class Xii Physics Assignment 1 Unit 1Document2 pagesClass Xii Physics Assignment 1 Unit 1vishal110085No ratings yet
- Markschemes FYE Sec 3Document2 pagesMarkschemes FYE Sec 3KonicNo ratings yet
- Quantum Physics For Beginners - A Comprehensive Guide For The StarterDocument319 pagesQuantum Physics For Beginners - A Comprehensive Guide For The StarterYiannis Thomaidis100% (6)
- MTC 336 Study Guide 1228594655Document3 pagesMTC 336 Study Guide 1228594655Aaron SotoNo ratings yet
- Difference Between Static Relays and Electromagnetic RelaysDocument2 pagesDifference Between Static Relays and Electromagnetic Relayskriitka86% (7)
- 39Document26 pages39Moamen MohamedNo ratings yet
- 19 - Dowel BarsDocument22 pages19 - Dowel BarsboyzesNo ratings yet
- January 2012 MS - Unit 4 Edexcel Chemistry A-LevelDocument24 pagesJanuary 2012 MS - Unit 4 Edexcel Chemistry A-LevelMaria KolokasiNo ratings yet
- 07Document3 pages07Ruswanto Ais ZalfaNo ratings yet
- FFT Zero PadDocument4 pagesFFT Zero PadKim Gerald TejadaNo ratings yet
- Reverse OsmosisDocument24 pagesReverse OsmosisHyumi DarthNo ratings yet
- Electromotive Force Magnetic FluxDocument8 pagesElectromotive Force Magnetic FluxJalal Al-SaifNo ratings yet