Download as pdf or txt
You might also like
- AP Statistics Chapter Notes (1-12)Document15 pagesAP Statistics Chapter Notes (1-12)Laura MesaNo ratings yet
- Lexus RX 350 RX 270 Wiring DiagramsDocument6 pagesLexus RX 350 RX 270 Wiring DiagramsPervaiz Anjum100% (1)
- Soundsystem DYNAUDIO®: Wiring DiagramDocument4 pagesSoundsystem DYNAUDIO®: Wiring DiagramHaji RashidNo ratings yet
- Gut Recovery Program: A New Approach To Treating Chronic Gastrointestinal InfectionsDocument47 pagesGut Recovery Program: A New Approach To Treating Chronic Gastrointestinal Infectionsautismone100% (4)
- Mitosis QuestionsDocument10 pagesMitosis QuestionscapremaeNo ratings yet
- Nominal Roll Cum Result Sheet - Certificate 'B' Exam: 2017Document16 pagesNominal Roll Cum Result Sheet - Certificate 'B' Exam: 2017Mari MuthuNo ratings yet
- VTU Updated Results After Revaluation 2022Document1 pageVTU Updated Results After Revaluation 2022mouneshjalli7353No ratings yet
- VTU Result 8th SemDocument1 pageVTU Result 8th SemDhanu DhanushNo ratings yet
- 2023 Army Builder 230101 - Mid Republican RomansDocument2 pages2023 Army Builder 230101 - Mid Republican RomansJustine QuiamcoNo ratings yet
- Audi Q2 No. 6 / 1: Airbag Systems, (L0L)Document9 pagesAudi Q2 No. 6 / 1: Airbag Systems, (L0L)acb . bNo ratings yet
- 2022 NCAA Women's Tournament Printable BracketDocument1 page2022 NCAA Women's Tournament Printable Bracketlamonica23No ratings yet
- Lc200 Stsrting SystemDocument6 pagesLc200 Stsrting SystemMichael Frank Vasques RenaciaNo ratings yet
- 12th ResDocument1 page12th Resvinay pratapNo ratings yet
- 24 Urban Cruiser (Cont. Next Page) : Abs (W/ VSC), TRC and VSCDocument2 pages24 Urban Cruiser (Cont. Next Page) : Abs (W/ VSC), TRC and VSCHEMIL ROBERTO RODRIGUEZ HERRERANo ratings yet
- VTU Result 2021 3rd SemDocument1 pageVTU Result 2021 3rd Semsteven iragarNo ratings yet
- Sui VTU ResultDocument2 pagesSui VTU ResultSantosh AngadiNo ratings yet
- BL B2 B2 B2 2 B2 (Htuin) : Qe Mhtoa Fh1 Sii L This Is To Certify That Yuvraj SetiaDocument1 pageBL B2 B2 B2 2 B2 (Htuin) : Qe Mhtoa Fh1 Sii L This Is To Certify That Yuvraj SetiaThe RealityNo ratings yet
- Vaccination Schedule Qatar Vs IndiaDocument1 pageVaccination Schedule Qatar Vs IndiaKiran Kumar AkulaNo ratings yet
- 2021 Ncaa Division I Men'S Basketball ChampionshipDocument1 page2021 Ncaa Division I Men'S Basketball ChampionshipJessica N.No ratings yet
- Aditya B.A All Yr ResultDocument1 pageAditya B.A All Yr ResultAJAY KUMAR GUPTANo ratings yet
- VTU Result PDFDocument2 pagesVTU Result PDFNaveen KumarNo ratings yet
- Twice 5th World Tour 'Ready To Be' Tickets Jul 02, 2023 Toronto, On TicketmasterDocument1 pageTwice 5th World Tour 'Ready To Be' Tickets Jul 02, 2023 Toronto, On Ticketmasterrezl9162No ratings yet
- 2021 NCAA Tournament BracketDocument1 page2021 NCAA Tournament BracketHouston ChronicleNo ratings yet
- TPMS Control Module WiringDocument1 pageTPMS Control Module Wiringcv01ssyNo ratings yet
- Cartas IAC Febrero 2020 Jeppesen PDFDocument323 pagesCartas IAC Febrero 2020 Jeppesen PDFOlivier Layly100% (1)
- ACW8516500 - C2 - Cabine MecânicoDocument13 pagesACW8516500 - C2 - Cabine MecânicoerriqueNo ratings yet
- Sony Vaio Pcg-61611 Quanta Ne7 Uma Da0ne7mb6d0 Rev DDocument42 pagesSony Vaio Pcg-61611 Quanta Ne7 Uma Da0ne7mb6d0 Rev DjuliocunachiNo ratings yet
- 1999 Ford M 1Document1 page1999 Ford M 1Rafa MackintoshNo ratings yet
- Grilla Semanal Julio SonyDocument5 pagesGrilla Semanal Julio Sonyapi-20000640No ratings yet
- m0043 003 1 PDFDocument1 pagem0043 003 1 PDFajarekarga ajarekarNo ratings yet
- Expedition 98 TIPO 3Document1 pageExpedition 98 TIPO 3Rogelio ArenasNo ratings yet
- Vote Intention JanuaryDocument5 pagesVote Intention JanuaryThe Globe and Mail100% (1)
- Connection: C021 Code: AISC 14th Edtion - LRFD)Document1 pageConnection: C021 Code: AISC 14th Edtion - LRFD)gv Sathishkumar KumarNo ratings yet
- Aits Dispatch Schedule: Distance Learning Programme Division (DLPD)Document1 pageAits Dispatch Schedule: Distance Learning Programme Division (DLPD)balaNo ratings yet
- Lane Change AssistDocument4 pagesLane Change AssistMartynas RamanauskasNo ratings yet
- Compass: Wiring DiagramDocument2 pagesCompass: Wiring DiagramHaji RashidNo ratings yet
- Compass: Wiring DiagramDocument2 pagesCompass: Wiring DiagramHaji RashidNo ratings yet
- Overall EWD Vehicle Interior Seat Belt Warning (RHD)Document4 pagesOverall EWD Vehicle Interior Seat Belt Warning (RHD)gabrielzinho43No ratings yet
- Overall EWD Vehicle Interior Combination Meter (RHD)Document4 pagesOverall EWD Vehicle Interior Combination Meter (RHD)gabrielzinho43No ratings yet
- ZF As Tronic Schematic E PDF PDF Transmission (Mechanics) VehiclesDocument1 pageZF As Tronic Schematic E PDF PDF Transmission (Mechanics) Vehiclesariswidi03No ratings yet
- Rosie Casals: WTA Finals Fort WorthDocument1 pageRosie Casals: WTA Finals Fort WorthHHPSHHNo ratings yet
- CREW: National Aeronautics and Space Administration: EUMETSAT Presentation To SUAGDocument17 pagesCREW: National Aeronautics and Space Administration: EUMETSAT Presentation To SUAGCREWNo ratings yet
- Engine Controls (Powertrain Management) - ALLDATA RepairDocument5 pagesEngine Controls (Powertrain Management) - ALLDATA RepairAUTOEPC LAAM (LUFACANA)No ratings yet
- Elt 345Document58 pagesElt 345miguel ayalaNo ratings yet
- SM 76Document4 pagesSM 76SuckmytickNo ratings yet
- View GraphicDocument1 pageView Graphicjohn gonzález camposNo ratings yet
- View GraphicDocument1 pageView Graphicjohn gonzález camposNo ratings yet
- View GraphicDocument1 pageView Graphicjohn gonzález camposNo ratings yet
- Computer Data LinesDocument1 pageComputer Data Linesluis dario mejia ocampo100% (1)
- Tamilnadu Teachers Education University, B.Ed./B.Ed (Special) - May/June 2010 (2009-2010) ResultsDocument4 pagesTamilnadu Teachers Education University, B.Ed./B.Ed (Special) - May/June 2010 (2009-2010) Resultsmmmmaran4uNo ratings yet
- New Old Noti 6th Sem Not212 215 Grade All Not Final 240122Document18 pagesNew Old Noti 6th Sem Not212 215 Grade All Not Final 240122Avni GuptaNo ratings yet
- 2022-2023 2022 10 30 12 46 55 Performance ReportDocument1 page2022-2023 2022 10 30 12 46 55 Performance ReportManoj ChauhanNo ratings yet
- Engine Controls (Powertrain Management) - ALLDATA RepairDocument5 pagesEngine Controls (Powertrain Management) - ALLDATA RepairAl final BuscoNo ratings yet
- Engine Controls (Powertrain Management) - ALLDATA RepairDocument5 pagesEngine Controls (Powertrain Management) - ALLDATA RepairAl final BuscoNo ratings yet
- 2015 Mazda CX-9 Sport 3.7LDocument81 pages2015 Mazda CX-9 Sport 3.7LData JuanNo ratings yet
- VTU Result 2023Document1 pageVTU Result 2023Kushal PNo ratings yet
- Bet9ja Nigeria Sport Betting,Premier League Odds,… 2Document1 pageBet9ja Nigeria Sport Betting,Premier League Odds,… 2kareemfemi53No ratings yet
- Cor002 - Stemh11 M3. 5Document7 pagesCor002 - Stemh11 M3. 5Joanna G JosonNo ratings yet
- 1 Column Setting Out, Rebar Cutting List For Column, SW & SFDocument6 pages1 Column Setting Out, Rebar Cutting List For Column, SW & SFCathy SaguindanNo ratings yet
- Direct-Hit - SearchDocument1 pageDirect-Hit - SearchCocatech StudiosNo ratings yet
- ಕ ಫ ಾಂಶ ಜೂ / ಜು ೖ - ೨೦೧೯. Vtu Provisional Results Of Ug / Pg June / July - 2019 ExaminationDocument1 pageಕ ಫ ಾಂಶ ಜೂ / ಜು ೖ - ೨೦೧೯. Vtu Provisional Results Of Ug / Pg June / July - 2019 ExaminationSantosh AngadiNo ratings yet
- 1.1 Disaster Managemen PHC-DM smt4 FKUBDocument48 pages1.1 Disaster Managemen PHC-DM smt4 FKUBrizqinapermatasariNo ratings yet
- Methodology - 2: Critical AppraisalDocument24 pagesMethodology - 2: Critical AppraisalrizqinapermatasariNo ratings yet
- Geriatric Syndromes: Dr. Sri Sunarti, SPPD K-Ger Divisi Geriatri Dan Gerontology Smf/Lab. Ipd Fkub/Rssa MalangDocument70 pagesGeriatric Syndromes: Dr. Sri Sunarti, SPPD K-Ger Divisi Geriatri Dan Gerontology Smf/Lab. Ipd Fkub/Rssa MalangrizqinapermatasariNo ratings yet
- Congenital Disorder - S1Document54 pagesCongenital Disorder - S1rizqinapermatasariNo ratings yet
- E Tra: Diagnosis and Management of Atopic Dermatitis: A ReviewDocument13 pagesE Tra: Diagnosis and Management of Atopic Dermatitis: A ReviewrizqinapermatasariNo ratings yet
- Management of Tonsillar Carcinoma With Advanced Radiation Therapy and Chemotherapy TechniquesDocument7 pagesManagement of Tonsillar Carcinoma With Advanced Radiation Therapy and Chemotherapy TechniquesrizqinapermatasariNo ratings yet
- Guidelines For Submitting Specimens To CDC For Laboratory Testing For Sars-Cov-2Document2 pagesGuidelines For Submitting Specimens To CDC For Laboratory Testing For Sars-Cov-2rizqinapermatasariNo ratings yet
- Pham Ngoc Thach University of Medicine DUOC2019Document109 pagesPham Ngoc Thach University of Medicine DUOC2019Nhung PhamNo ratings yet
- bt101 (Grey Format)Document379 pagesbt101 (Grey Format)Hasnain HaiderNo ratings yet
- Chaos Theory, Evolution and The CreationDocument39 pagesChaos Theory, Evolution and The CreationDennis MurphyNo ratings yet
- Zoo Lab Frog ActivityDocument12 pagesZoo Lab Frog ActivityAdznaira AmilussinNo ratings yet
- Virus Web WorksheetDocument3 pagesVirus Web WorksheetBrian Ramirez RamirezNo ratings yet
- Full Ebook of Physical Exercise and Natural and Synthetic Products in Health and Disease Paul C Guest Online PDF All ChapterDocument69 pagesFull Ebook of Physical Exercise and Natural and Synthetic Products in Health and Disease Paul C Guest Online PDF All Chapteranthonybrister74480100% (7)
- Cell Parts and FunctionDocument4 pagesCell Parts and FunctionGem CarpioNo ratings yet
- Good Food Label Worksheet - Angie 2012Document3 pagesGood Food Label Worksheet - Angie 2012Dwi SuprionoNo ratings yet
- EngineeringDocument105 pagesEngineeringKrishan Kumar SinghNo ratings yet
- 13 Quarter 1 Module 13-INTRODUCTION-TO-PHOTOSYNTHESISDocument16 pages13 Quarter 1 Module 13-INTRODUCTION-TO-PHOTOSYNTHESISMah Jane Divina100% (1)
- Introduction To NeuroanatomyDocument49 pagesIntroduction To NeuroanatomyPratik Dhakal100% (1)
- Edquist 1997Document44 pagesEdquist 1997Digamber SinghNo ratings yet
- ArticuloDocument7 pagesArticuloCarlosGuevaraPáezNo ratings yet
- Science Continuous Assessment (Chapter 1 & 2)Document10 pagesScience Continuous Assessment (Chapter 1 & 2)Catherine ChanNo ratings yet
- Reading Comprehension: HIVDocument2 pagesReading Comprehension: HIVFlori75% (8)
- Lymphatic System OutlineDocument4 pagesLymphatic System OutlineMelljonhNo ratings yet
- Stoichiometry of Microbial Growth and Product FormationDocument10 pagesStoichiometry of Microbial Growth and Product FormationfayeNo ratings yet
- MM Protein Synthesis Activity CBdiTEEDocument6 pagesMM Protein Synthesis Activity CBdiTEESHARIFAH BINTI HASSAN MoeNo ratings yet
- Anatomy and Physiology ReviewerDocument17 pagesAnatomy and Physiology ReviewerJanel EustaquioNo ratings yet
- Virus-Klasifikasi, Sifat, GenetikaDocument26 pagesVirus-Klasifikasi, Sifat, Genetikanovia putriNo ratings yet
- Diagnostic and Microbiological Tools in Oral and Maxillofacial SurgeryDocument36 pagesDiagnostic and Microbiological Tools in Oral and Maxillofacial SurgeryrajtanniruNo ratings yet
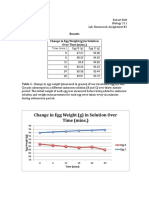
- Results Change in Egg Weight (G) in Solution Over Time (Mins.)Document2 pagesResults Change in Egg Weight (G) in Solution Over Time (Mins.)Roberto BeltNo ratings yet
- Function of Thyroid Hormone: Muhammad Wajid Institute of PharmacyDocument14 pagesFunction of Thyroid Hormone: Muhammad Wajid Institute of PharmacyAhmed ImranNo ratings yet
- MetaboNews Aug2022Document15 pagesMetaboNews Aug2022Miguel Fernandez GarciaNo ratings yet
- TOPIC 6.1 - 6.2 - DNA Structure and Replication, Student Learning GuideDocument4 pagesTOPIC 6.1 - 6.2 - DNA Structure and Replication, Student Learning GuideL ChanNo ratings yet
- The Genetic Relation of Indonesian Calloselasma Rhodostoma Based On Nd4 Gene and Preliminary Study 7941Document8 pagesThe Genetic Relation of Indonesian Calloselasma Rhodostoma Based On Nd4 Gene and Preliminary Study 7941Ichda Arini DinanaNo ratings yet
- (Labgenomics) Biz Collaboration - LabGun™ COVID-19 Assay PCR Kit (Information 20200304) - ENDocument15 pages(Labgenomics) Biz Collaboration - LabGun™ COVID-19 Assay PCR Kit (Information 20200304) - ENJoana SampaioNo ratings yet
- Anthropology Wbcs Main Xam PapersDocument33 pagesAnthropology Wbcs Main Xam PapersMainak SasmalNo ratings yet