Download as pdf or txt
You might also like
- Braddom 6th CH 47 Cerebral PalsyDocument34 pagesBraddom 6th CH 47 Cerebral Palsy李宗旻No ratings yet
- Anterolisthesis - Symptoms, Causes and Treatment PDFDocument6 pagesAnterolisthesis - Symptoms, Causes and Treatment PDFRiris MayasariNo ratings yet
- Tumor Inhibitors From PlantsDocument5 pagesTumor Inhibitors From PlantsMuhammad Mustafa Ijaz0% (1)
- A Schematic Approach To Hypotonia in Infancy: Une Démarche Schématique Envers L'hypotonie Pendant La Première EnfanceDocument4 pagesA Schematic Approach To Hypotonia in Infancy: Une Démarche Schématique Envers L'hypotonie Pendant La Première EnfanceRahul RaiNo ratings yet
- Cerebral Palsy An Overview 2018Document11 pagesCerebral Palsy An Overview 2018Yhuliana AcostaNo ratings yet
- Kuliah OiDocument61 pagesKuliah OiNovasuryatiNo ratings yet
- Osteogenesis ImperfectaDocument27 pagesOsteogenesis ImperfectaAlejandro Reyes50% (2)
- Osteogenesis Imperfecta:: A Guide For Medical Professionals, Individuals, and Families Affected by OIDocument14 pagesOsteogenesis Imperfecta:: A Guide For Medical Professionals, Individuals, and Families Affected by OIGemma GarciaNo ratings yet
- Diseases of Bone MangalaDocument246 pagesDiseases of Bone MangalaharshiniNo ratings yet
- Osteogenisis Imperfecta: Christopher LimDocument34 pagesOsteogenisis Imperfecta: Christopher LimRhomadhoni Ika PutraNo ratings yet
- Osteogenesis Imperfecta:: Primary CareDocument13 pagesOsteogenesis Imperfecta:: Primary CareLala AkamatsuNo ratings yet
- Low Birth WeightDocument2 pagesLow Birth WeightDjfnswn UkhtyNo ratings yet
- The Following Are Types of Osteogenesis Imperfecta and Their SymptomsDocument4 pagesThe Following Are Types of Osteogenesis Imperfecta and Their SymptomsPerlyn EndozoNo ratings yet
- Unusual Association of Meningioma and Pituitary Hypoplasia in A Patient With Osteogenesis ImperfectaDocument4 pagesUnusual Association of Meningioma and Pituitary Hypoplasia in A Patient With Osteogenesis ImperfectaIJAR JOURNALNo ratings yet
- Medicina: Current Overview of Osteogenesis ImperfectaDocument15 pagesMedicina: Current Overview of Osteogenesis ImperfectaCramer IonelaNo ratings yet
- Physiotherapy in The Management of Osteogenesis Imperfecta 2Document38 pagesPhysiotherapy in The Management of Osteogenesis Imperfecta 2Fatima Habbaba JibrinNo ratings yet
- Testing For Osteogenesis ImperfectaDocument5 pagesTesting For Osteogenesis ImperfectareinzluiNo ratings yet
- Osteogenesis ImperfectaDocument8 pagesOsteogenesis ImperfectaClariss AlotaNo ratings yet
- Osteogenesis ImperfectaDocument6 pagesOsteogenesis Imperfectamiss RN100% (2)
- Cerebral Palsy: MBBS YEAR 3, Batch 8, 2019 DR - Swe Zin Aye Zinaye - Swe@qiup - Edu.myDocument37 pagesCerebral Palsy: MBBS YEAR 3, Batch 8, 2019 DR - Swe Zin Aye Zinaye - Swe@qiup - Edu.myVishalli KalaiwananNo ratings yet
- Paediatric Clerkship 1Document9 pagesPaediatric Clerkship 1fredrick damian100% (1)
- Hypotonia: A Clinical Sign, Different Etiologies: Keywords: Infant Newborn Muscle Hypotonia/etiologyDocument6 pagesHypotonia: A Clinical Sign, Different Etiologies: Keywords: Infant Newborn Muscle Hypotonia/etiologyAna SopaNo ratings yet
- Recurrent FracturesDocument11 pagesRecurrent FracturesSohaila GodaNo ratings yet
- Journal Osteogenesis ImperfectaDocument7 pagesJournal Osteogenesis ImperfectaMatheis Laskar PelangiNo ratings yet
- تمريض الاطفال نظري 6Document31 pagesتمريض الاطفال نظري 6jadarcNo ratings yet
- 00 Paediatric Clerkship-1Document9 pages00 Paediatric Clerkship-1Lexis SanchizieNo ratings yet
- 1.nutritional Disorder With KeyDocument9 pages1.nutritional Disorder With KeykarthicpraveenNo ratings yet
- Osteogenesis ImperfectaDocument11 pagesOsteogenesis ImperfectaDokter ZukieNo ratings yet
- Case Reflection 1 POD 1Document7 pagesCase Reflection 1 POD 1seankelly327No ratings yet
- Cerebral Palsy Lecture1 ٠٥١٤٥٠Document56 pagesCerebral Palsy Lecture1 ٠٥١٤٥٠nagmxxmNo ratings yet
- 2023 IBCLC Detailed Content Outline FINALDocument5 pages2023 IBCLC Detailed Content Outline FINALDilmaNo ratings yet
- A Rare Cause of Pancreatic Insufficiency Johanson Blizzard SyndromeDocument3 pagesA Rare Cause of Pancreatic Insufficiency Johanson Blizzard Syndromeد نبيل عبيدNo ratings yet
- A Case Report: Osteogenesis ImperfectaDocument27 pagesA Case Report: Osteogenesis ImperfectaJardee DatsimaNo ratings yet
- Leg Bowing 1Document42 pagesLeg Bowing 1Mitulsinh M RavaljiNo ratings yet
- Osteogenesis Imperfecta Synonyms and Related Keywords: Osteogenesis Imperfecta, OI, Fragile Bone Disease, BrittleDocument14 pagesOsteogenesis Imperfecta Synonyms and Related Keywords: Osteogenesis Imperfecta, OI, Fragile Bone Disease, BrittledhawnmikiNo ratings yet
- Notes On Spinal DysraphismDocument3 pagesNotes On Spinal DysraphismMicaela GarciaNo ratings yet
- Bloom SyndromeDocument4 pagesBloom SyndromeMeryam MeryNo ratings yet
- Osteogenesis Imperfecta Written ReportDocument16 pagesOsteogenesis Imperfecta Written Reportkimchi girlNo ratings yet
- Stone Man Syndrome (Shini)Document12 pagesStone Man Syndrome (Shini)HarshiniNo ratings yet
- 2009 Article 214Document9 pages2009 Article 214Lema Ulloa ZoraidaNo ratings yet
- Physeal Closure Time & DiseaseDocument8 pagesPhyseal Closure Time & DiseaseMelissa StrawhunNo ratings yet
- Osteogenesis Imperfecta (OI and Sometimes Known As Brittle Bone DiseaseDocument4 pagesOsteogenesis Imperfecta (OI and Sometimes Known As Brittle Bone Diseaseadrielle27No ratings yet
- CNS 2Document9 pagesCNS 2husainozelNo ratings yet
- Skeletal Abnormalities of The Upper Limbs - Neonatal Diagnosis of 49XXXXY SyndromeDocument4 pagesSkeletal Abnormalities of The Upper Limbs - Neonatal Diagnosis of 49XXXXY SyndromeFelipe TavaresNo ratings yet
- Prognóstico para CaminharDocument6 pagesPrognóstico para CaminharWenderson MoraisNo ratings yet
- 10 Diesease of Infancy and ChildhoodDocument31 pages10 Diesease of Infancy and ChildhoodRholter Dave LeeNo ratings yet
- Cozzolino 2016Document7 pagesCozzolino 2016Shinohara YuusukeNo ratings yet
- Dismorfico en Talla Baja AdultosDocument6 pagesDismorfico en Talla Baja AdultosEliana Orozco HenaoNo ratings yet
- Positional Plagiocephaly From Structure To Function Clinic - 2020 - Early HumanDocument6 pagesPositional Plagiocephaly From Structure To Function Clinic - 2020 - Early HumanmireliNo ratings yet
- Paediatrics - Child With Basic Nutrition NeedDocument13 pagesPaediatrics - Child With Basic Nutrition NeedhalesNo ratings yet
- Uas 8 - TeratologiDocument44 pagesUas 8 - TeratologiRamdinArsitekNo ratings yet
- Pediatric Usmle NotesDocument14 pagesPediatric Usmle NotesLucykesh67% (3)
- 3RD TERM JSS2 PHE LESSON NOTES UsuavsDocument19 pages3RD TERM JSS2 PHE LESSON NOTES UsuavsIdeji EkemenaNo ratings yet
- Mental RetardationDocument23 pagesMental Retardation5xqqq8mx6pNo ratings yet
- SDL 1Document8 pagesSDL 1Jona Joyce JunsayNo ratings yet
- Cerebral PalsyDocument97 pagesCerebral PalsyPrernaSharma100% (3)
- Congential 3 Musculoskeletal 3 Neurological 3 Abusive DisordersDocument7 pagesCongential 3 Musculoskeletal 3 Neurological 3 Abusive DisordersNichole CollinsNo ratings yet
- Skeletal Manifestations of Scurvy - Case Report DubaiDocument5 pagesSkeletal Manifestations of Scurvy - Case Report DubaigistaluvikaNo ratings yet
- Cerebral Palsy: Submitted By: Prerna Sharma M.SC Nursing, 4 SemesterDocument97 pagesCerebral Palsy: Submitted By: Prerna Sharma M.SC Nursing, 4 Semestervarshasharma05100% (2)
- 31.bone & Joint DisordersDocument128 pages31.bone & Joint DisordersSulistyawati WrimunNo ratings yet
- Floppy InfantDocument5 pagesFloppy InfantRajesh pooniaNo ratings yet
- Fibrodysplasia Ossifican Progressiva (Stone Man Syndrome), A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsFrom EverandFibrodysplasia Ossifican Progressiva (Stone Man Syndrome), A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsNo ratings yet
- Draft Skripsi TasyaDocument108 pagesDraft Skripsi TasyaErlitha Azhari DewiNo ratings yet
- Periode: 01/06/2021 25/06 (WARKUR) Catatan: Lengkapi Dengan Manual Bagian Warna OrangeDocument34 pagesPeriode: 01/06/2021 25/06 (WARKUR) Catatan: Lengkapi Dengan Manual Bagian Warna OrangeErlitha Azhari DewiNo ratings yet
- 04 JASPEL Non Kpts WARKUR BLN MARET 20Document25 pages04 JASPEL Non Kpts WARKUR BLN MARET 20Erlitha Azhari DewiNo ratings yet
- Update On The Management of HypophosphatasiaDocument8 pagesUpdate On The Management of HypophosphatasiaErlitha Azhari DewiNo ratings yet
- Implementation of Pathogen Reduction Technology in The Manufacture of Blood Components in Blood Establishments Questions and Answers Draft Guidance For IndustryDocument14 pagesImplementation of Pathogen Reduction Technology in The Manufacture of Blood Components in Blood Establishments Questions and Answers Draft Guidance For Industrydrsapta manNo ratings yet
- Pediatric Nursing Dissertation TopicsDocument8 pagesPediatric Nursing Dissertation TopicsCustomCollegePaperCanada100% (1)
- Implementing Injury Prevention: The Rocky Road From RCT To Real-World Injury ReductionDocument7 pagesImplementing Injury Prevention: The Rocky Road From RCT To Real-World Injury ReductionAbdul Wahid ShaikhNo ratings yet
- Shortened Dental ArchDocument9 pagesShortened Dental ArchSanjay HirekodiNo ratings yet
- Hemolytic Disease of NewbornDocument41 pagesHemolytic Disease of NewbornRaja100% (3)
- Rajsthan All Surgical Distributor Data ListDocument33 pagesRajsthan All Surgical Distributor Data ListcatalogueNo ratings yet
- Management of Genital ProlapseDocument39 pagesManagement of Genital Prolapsecool_ice2t6No ratings yet
- Case Scenario/Situation Answer/Explanation: NCM 101 RLE - Health AssessmentDocument2 pagesCase Scenario/Situation Answer/Explanation: NCM 101 RLE - Health AssessmentmaryNo ratings yet
- Cultural Humility Versus Cultural Competence - A Critical Distinction in Defining Physician Training Outcomes in Multicultural Education (3) - 0Document10 pagesCultural Humility Versus Cultural Competence - A Critical Distinction in Defining Physician Training Outcomes in Multicultural Education (3) - 0Gabriela BarreiraNo ratings yet
- Effectiveness of Anterior Repositioning Orthotic Splints in Recapturing The TMJ DiscDocument10 pagesEffectiveness of Anterior Repositioning Orthotic Splints in Recapturing The TMJ DiscRyan SperzelNo ratings yet
- Apical Periodontitis Treatment Surgical-Non SurgicDocument1 pageApical Periodontitis Treatment Surgical-Non SurgicTatiana DimaNo ratings yet
- 1st Quarter 1 CONSUMER HEALTH EDUCATION (Autosaved)Document16 pages1st Quarter 1 CONSUMER HEALTH EDUCATION (Autosaved)JEANY BARROZONo ratings yet
- Report PDFDocument6 pagesReport PDFAayushiNo ratings yet
- Common Neonatal ProblemsDocument32 pagesCommon Neonatal ProblemsbrhomzalatNo ratings yet
- Precision in MSDocument62 pagesPrecision in MSSnezana MihajlovicNo ratings yet
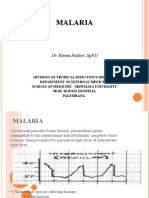
- Malaria: Dr. Harun Hudari, SPPDDocument49 pagesMalaria: Dr. Harun Hudari, SPPDEdvans HenryNo ratings yet
- Test Bank For Pathophysiology 6th Edition Jacquelyn L BanasikDocument5 pagesTest Bank For Pathophysiology 6th Edition Jacquelyn L BanasikJesus Carter97% (38)
- Gouty Arthritis: Tarlac State University College of Science Nursing DepartmentDocument22 pagesGouty Arthritis: Tarlac State University College of Science Nursing DepartmentKrisianne Mae Lorenzo FranciscoNo ratings yet
- IVM (In Vitro Maturation) WWWWWWWWWDocument17 pagesIVM (In Vitro Maturation) WWWWWWWWWMirza HassanNo ratings yet
- Badac Plan TunasanDocument2 pagesBadac Plan TunasanjonahNo ratings yet
- PDFDocument11 pagesPDFNico IonaşcuNo ratings yet
- The Impact of Quality-Reporting Programs On Hospital OperationsDocument12 pagesThe Impact of Quality-Reporting Programs On Hospital OperationsVipul GuptaNo ratings yet
- Siddha Applied Science InstituteDocument4 pagesSiddha Applied Science InstituteLagin DranNo ratings yet
- Organisational Structure - Asian HeartDocument13 pagesOrganisational Structure - Asian HeartshwetadhotarNo ratings yet
- Moot Court BrochureDocument16 pagesMoot Court BrochureAkashNo ratings yet
- Pharmacoeconomics: BY Mrs. K.Shailaja., M. Pharm., Lecturer Dept of Pharmacy Practice, SRM College of PharmacyDocument15 pagesPharmacoeconomics: BY Mrs. K.Shailaja., M. Pharm., Lecturer Dept of Pharmacy Practice, SRM College of Pharmacytsegaab yosephNo ratings yet
- Nephrotic SyndromeDocument84 pagesNephrotic SyndromeRahul DhakerNo ratings yet
- What You Need To Know About Sleep Apnea: Statement of RightsDocument27 pagesWhat You Need To Know About Sleep Apnea: Statement of Rightslnln462lnNo ratings yet