
Turkevich1985 Article ColloidalGoldPartII PDF
Turkevich1985 Article ColloidalGoldPartII PDF
You might also like
- Chem Lab Report - Electronic Absorption Spectra of Some Cu ComplexesDocument6 pagesChem Lab Report - Electronic Absorption Spectra of Some Cu ComplexesMiguel Ackah-Yensu93% (14)
- XRD Lab ReportDocument3 pagesXRD Lab ReportArman Boroomand67% (3)
- en 12004-2-2017 CenDocument35 pagesen 12004-2-2017 CenMalak Hindi100% (2)
- This Study Resource Was: SOILS 101 Chapter 5 AssignmentDocument5 pagesThis Study Resource Was: SOILS 101 Chapter 5 AssignmentSunil DhankharNo ratings yet
- Ligand Field StrengthDocument24 pagesLigand Field StrengthIrvandar NurviandyNo ratings yet
- CFT PDFDocument20 pagesCFT PDFRUFAS KANIKANTINo ratings yet
- CBSE Class 12 Chemistry Paper Sample Paper Solution Set 4Document14 pagesCBSE Class 12 Chemistry Paper Sample Paper Solution Set 4Gamer ChannelNo ratings yet
- Atomic Structure (1-35)Document35 pagesAtomic Structure (1-35)deepakkr08075% (4)
- Optical Properties of Metal Nanoparticles: Sriharsha KarumuriDocument12 pagesOptical Properties of Metal Nanoparticles: Sriharsha Karumurigea1616No ratings yet
- Interpretation of The Emission Spectra of Trivalent Chromium-Doped Garnet Crystals Using Tanabe-Sugano DiagramsDocument3 pagesInterpretation of The Emission Spectra of Trivalent Chromium-Doped Garnet Crystals Using Tanabe-Sugano DiagramsDaniela Araújo RodríguezNo ratings yet
- Transition Metal 4Document4 pagesTransition Metal 4Sushant ShahNo ratings yet
- Chapter 3Document47 pagesChapter 3蘇翊愷No ratings yet
- Monolayer-Protected Metal Nanoparticle: Toluene) Auc L Toluene) N (C H Auc L N (C HDocument27 pagesMonolayer-Protected Metal Nanoparticle: Toluene) Auc L Toluene) N (C H Auc L N (C HmichsantosNo ratings yet
- Color of Transition Metal IonsDocument19 pagesColor of Transition Metal IonsSyedah Maira ShahNo ratings yet
- (13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsDocument10 pages(13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsVikash KumarNo ratings yet
- Structure of Atom Class 11 Chemistry CBSE BoardDocument5 pagesStructure of Atom Class 11 Chemistry CBSE BoardminimataNo ratings yet
- Structure of Atom: Sub-Atomic Particles: Name Symbol Charge/C Relative Charge Mass/kgDocument8 pagesStructure of Atom: Sub-Atomic Particles: Name Symbol Charge/C Relative Charge Mass/kgSparsh MehtaNo ratings yet
- Solid State ChemistryDocument36 pagesSolid State ChemistrySoumya BullaNo ratings yet
- Chapter 2 - Structure of AtomDocument18 pagesChapter 2 - Structure of AtomstudyforiittomeetbtsNo ratings yet
- I. 1. What Are Nanoparticles?: Figure 1. Dimension of Different Objects in The Length ScaleDocument11 pagesI. 1. What Are Nanoparticles?: Figure 1. Dimension of Different Objects in The Length ScaleAmandeep Singh PannuNo ratings yet
- S/Sciences/Physics/Optics/Lasertut Orial/Creating/Creating - HTMDocument13 pagesS/Sciences/Physics/Optics/Lasertut Orial/Creating/Creating - HTMSuvankar ChakrabortyNo ratings yet
- CH 2 Structure of Atom PDFDocument18 pagesCH 2 Structure of Atom PDFjazi_4uNo ratings yet
- Grain Boundaries in MetalsDocument2 pagesGrain Boundaries in MetalsAvanish KumarNo ratings yet
- Atomic StructureDocument10 pagesAtomic StructureSatyam MittalNo ratings yet
- D and F BlockDocument8 pagesD and F BlockAnanyaNo ratings yet
- Lecture 2 CY101Document21 pagesLecture 2 CY101Gyanaranjan SahooNo ratings yet
- 11 Chemistry Notes Ch02 Structure of AtomDocument18 pages11 Chemistry Notes Ch02 Structure of Atomyuvrajbrand01No ratings yet
- Teori Meda KristalDocument41 pagesTeori Meda KristalGilang Maulana PutraNo ratings yet
- 2008 Tuning Luminescence Intensity of RHO6G Dye Using SilverDocument4 pages2008 Tuning Luminescence Intensity of RHO6G Dye Using Silverantoich antoichNo ratings yet
- Bonding Models in Inorganic Chemistry 1. Ionic CompoundsDocument12 pagesBonding Models in Inorganic Chemistry 1. Ionic Compoundsfavourabiola123No ratings yet
- 11-CHEMISTRY (CH-2) PART-1Document3 pages11-CHEMISTRY (CH-2) PART-1mt3835570No ratings yet
- Structure of AtomDocument72 pagesStructure of AtomAditi YadavNo ratings yet
- Chapter 12 - Atoms-Saju-Hsslive PDFDocument9 pagesChapter 12 - Atoms-Saju-Hsslive PDFAmiNo ratings yet
- Raman Scattering: Jordan University of Science and TechnologyDocument40 pagesRaman Scattering: Jordan University of Science and TechnologyhaimantiNo ratings yet
- Crystal Field Theory - NURDocument5 pagesCrystal Field Theory - NURNurhajrahNo ratings yet
- Initial Oxidation Rate of Metals and The Logarithmic EquationDocument14 pagesInitial Oxidation Rate of Metals and The Logarithmic EquationAndrea CalderaNo ratings yet
- 11 Chemistry Notes Ch02 Structure of AtomDocument18 pages11 Chemistry Notes Ch02 Structure of AtomSayantanBanerjee0% (1)
- Co Ordination 1Document33 pagesCo Ordination 1Nivi RajNo ratings yet
- Physicsatoms 46198Document11 pagesPhysicsatoms 46198user 003No ratings yet
- Atomic Structure PDFDocument40 pagesAtomic Structure PDFAnkit MaanNo ratings yet
- Chemistry Form 6 Sem 2 03Document45 pagesChemistry Form 6 Sem 2 03Ng Swee Loong StevenNo ratings yet
- Structure of Atom - 404Document72 pagesStructure of Atom - 404vipulvidhya2020No ratings yet
- Structure of AtomDocument8 pagesStructure of AtompreetikoravNo ratings yet
- CHM 112Document16 pagesCHM 112olamideolafimihan040No ratings yet
- Permittivity and Transmission of MetalsDocument3 pagesPermittivity and Transmission of MetalsmaxNo ratings yet
- Iit - JEE Syllabus: RSM79 PH I AS CH 1Document48 pagesIit - JEE Syllabus: RSM79 PH I AS CH 1MD IMRAN100% (1)
- 2.structure of Atom-Smart Booklet-1Document44 pages2.structure of Atom-Smart Booklet-1nadeemnagthan008No ratings yet
- Inorganic Chem 1 2 PDFDocument73 pagesInorganic Chem 1 2 PDFYT ChongNo ratings yet
- PNGE352 - Lec3 - GR Logs-April1a - 2022Document22 pagesPNGE352 - Lec3 - GR Logs-April1a - 2022adnan.bunyNo ratings yet
- Structure of Atom Class 11Document74 pagesStructure of Atom Class 11Komal VermaNo ratings yet
- Energy Level SplittingDocument4 pagesEnergy Level SplittingMa'arif A. SyafiiNo ratings yet
- DFFHMDocument9 pagesDFFHMyaswanthNo ratings yet
- Atoms and NucleiDocument66 pagesAtoms and NucleiphysicsbypaulsirNo ratings yet
- Atomic and Nuclear Physics: Thomson's Atomic ModelDocument23 pagesAtomic and Nuclear Physics: Thomson's Atomic ModelDHINESH HARIKUMARNo ratings yet
- Atoms:: Particle Electron Proton Neutron Discovery Nature of Charge Negative Amount of Charge MassDocument6 pagesAtoms:: Particle Electron Proton Neutron Discovery Nature of Charge Negative Amount of Charge MassNasser SsennogaNo ratings yet
- Atomic NucleusDocument15 pagesAtomic Nucleussreenivas1990100% (1)
- Amorphous Semiconductors: Structural, Optical, and Electronic PropertiesFrom EverandAmorphous Semiconductors: Structural, Optical, and Electronic PropertiesNo ratings yet
- Mechanism of Organic ReactionDocument4 pagesMechanism of Organic ReactionRSLNo ratings yet
- Colloidal Gold. Part I: Historical and Preparative Aspects, Morphology and StructureDocument6 pagesColloidal Gold. Part I: Historical and Preparative Aspects, Morphology and StructureRSL100% (1)
- Carbon Monoxide or Carbonyl: MO DescriptionDocument3 pagesCarbon Monoxide or Carbonyl: MO DescriptionRSLNo ratings yet
- NH4BH4Document1 pageNH4BH4RSLNo ratings yet
- Coordination Isomers ListDocument1 pageCoordination Isomers ListRSLNo ratings yet
- Basics of ElectrochemistryDocument22 pagesBasics of ElectrochemistryRSLNo ratings yet
- DebereinerDocument4 pagesDebereinerRSLNo ratings yet
- S-Block Elments: Inorganic ChemistryDocument8 pagesS-Block Elments: Inorganic ChemistryRSLNo ratings yet
- Epoxides Ring-Opening - Chemistry LibreTextsDocument3 pagesEpoxides Ring-Opening - Chemistry LibreTextsRSLNo ratings yet
- © 1934 Nature Publishing GroupDocument2 pages© 1934 Nature Publishing GroupRSLNo ratings yet
- Colloids: Thomas Graham (1861) Studied The Ability of Dissolved Substances ToDocument28 pagesColloids: Thomas Graham (1861) Studied The Ability of Dissolved Substances ToRSLNo ratings yet
- List of Straight-Chain AlkanesDocument6 pagesList of Straight-Chain AlkanesRSLNo ratings yet
- Piezoelectric Materials: Crystal Orientation and Poling Direction - COMSOL Blog PDFDocument4 pagesPiezoelectric Materials: Crystal Orientation and Poling Direction - COMSOL Blog PDFRSLNo ratings yet
- Priority List IUPACDocument1 pagePriority List IUPACRSLNo ratings yet
- 2019dec-03 - Ionic Equilibrium - PracticeSheetDocument2 pages2019dec-03 - Ionic Equilibrium - PracticeSheetRSLNo ratings yet
- Redox Reactions and Balancing Using Oxidation Number & NfactorDocument1 pageRedox Reactions and Balancing Using Oxidation Number & NfactorRSLNo ratings yet
- Analysis Report of Criminal and Financial Background Details of Winners in Lok Sabha 2019 ElectionsDocument397 pagesAnalysis Report of Criminal and Financial Background Details of Winners in Lok Sabha 2019 ElectionsRSLNo ratings yet
- Colligative Properties: Cryoscopy & EbulliosDocument30 pagesColligative Properties: Cryoscopy & EbulliosRSL100% (1)
- Ionic Equilibrium - Salt Hydrolysis Practice Questions (Level-1)Document3 pagesIonic Equilibrium - Salt Hydrolysis Practice Questions (Level-1)RSLNo ratings yet
- Isoelectric Point, PiDocument2 pagesIsoelectric Point, PiRSLNo ratings yet
- EAS Worksheet 1 SubjDocument1 pageEAS Worksheet 1 SubjRSLNo ratings yet
- Koopmans TheoremDocument14 pagesKoopmans TheoremRSLNo ratings yet
- The World of Chemistry Episode 5 - A Matter of StateDocument2 pagesThe World of Chemistry Episode 5 - A Matter of StateRSL0% (1)
- Chemistry Organic GOC Reaction Mechanism Elimination E1 E2 E1cBDocument4 pagesChemistry Organic GOC Reaction Mechanism Elimination E1 E2 E1cBRSL100% (1)
- Crossword Coordination CompoundsDocument2 pagesCrossword Coordination CompoundsRSLNo ratings yet
- Factors Affecting Relative Strengths of Acids and BasesDocument1 pageFactors Affecting Relative Strengths of Acids and BasesRSL80% (5)
- Fibre Epoxy Composites at Low Temperature HartwigDocument9 pagesFibre Epoxy Composites at Low Temperature HartwigodormicchiNo ratings yet
- Handbook of Advanced Plasma Processing Techniques Book ReviewDocument1 pageHandbook of Advanced Plasma Processing Techniques Book Reviewmustafa alasadyNo ratings yet
- Proofex GPXDocument2 pagesProofex GPXVenkata RaoNo ratings yet
- Rocky 3 Technical ManualDocument8 pagesRocky 3 Technical ManualMáximo ValdésNo ratings yet
- Measuring Adhesion by Tape Test: Standard Test Methods ForDocument7 pagesMeasuring Adhesion by Tape Test: Standard Test Methods ForJony NavaNo ratings yet
- CE 321 Chapter 7 Express For PDFDocument43 pagesCE 321 Chapter 7 Express For PDFGerald MoaresNo ratings yet
- Principles of AdhesionDocument44 pagesPrinciples of AdhesionOmar Hesham SalahNo ratings yet
- NRCC 53261Document18 pagesNRCC 53261MEHDI FARROKHINo ratings yet
- An Alternative Method For Thermal Cycling Test - Effect On The Marginal Microleakage and Bond Strength of Dental Polymer Bonded To Dentin PDFDocument5 pagesAn Alternative Method For Thermal Cycling Test - Effect On The Marginal Microleakage and Bond Strength of Dental Polymer Bonded To Dentin PDFilich sevillaNo ratings yet
- Kinetic Molecular Model of Solids and Liquids Activity 1: Color DropDocument12 pagesKinetic Molecular Model of Solids and Liquids Activity 1: Color DropElaine Mae G. EsqueroNo ratings yet
- Concepts of Surface TensionDocument31 pagesConcepts of Surface TensionMartin AdriazolaNo ratings yet
- BS 3900 Pull Off Test PDFDocument9 pagesBS 3900 Pull Off Test PDFAvinash LalNo ratings yet
- Particle Size Effect On The Hydrophobicity and The Natural Floatability of MolybdeniteDocument105 pagesParticle Size Effect On The Hydrophobicity and The Natural Floatability of MolybdeniteAgustín F. Correa100% (1)
- Bitumen Binder and TypesDocument47 pagesBitumen Binder and TypesTarun Goel50% (2)
- Keur 00824-401 201505Document4 pagesKeur 00824-401 201505erik0007No ratings yet
- Proofex 12 PDFDocument2 pagesProofex 12 PDFmilanbrasinaNo ratings yet
- 2023 - Gordon - Surface Enineering Polymeric Materials Towards Lunar Dust MitigationDocument6 pages2023 - Gordon - Surface Enineering Polymeric Materials Towards Lunar Dust Mitigationjames.millsNo ratings yet
- Contact Angles: Surface TensionDocument4 pagesContact Angles: Surface TensionArpan GhoshNo ratings yet
- 1 Corrosion Prevention by Protective Coatings (Munger)Document19 pages1 Corrosion Prevention by Protective Coatings (Munger)Gagan MehandirattaNo ratings yet
- A006 Pi VtmoeoDocument3 pagesA006 Pi VtmoeoRick LeeKang WangNo ratings yet
- D 1779 - 98 R04 Rde3nzk - PDFDocument3 pagesD 1779 - 98 R04 Rde3nzk - PDFMarceloNo ratings yet
- Astm D7234-19Document9 pagesAstm D7234-19AhmedNo ratings yet
- The Potential of Using Agricultural Waste: Corn Husk For Particleboard Raw MaterialDocument9 pagesThe Potential of Using Agricultural Waste: Corn Husk For Particleboard Raw MaterialDessy BelmonteNo ratings yet
- Inks For Printing On Plastic FilmsDocument8 pagesInks For Printing On Plastic FilmsLucas Del PretteNo ratings yet
- ASTM D429 Rubber To Metal Adhesion Test EquipmentDocument4 pagesASTM D429 Rubber To Metal Adhesion Test EquipmentLimNo ratings yet
- Waste Plastic Modified BituminDocument11 pagesWaste Plastic Modified BituminMd.imthiyazNo ratings yet
- Plasmatreat ImagebrochureDocument28 pagesPlasmatreat ImagebrochurechibigarNo ratings yet
- GSECL Vidyut Sahayak Junior Engineer - Environment 18-02-2018 PDFDocument20 pagesGSECL Vidyut Sahayak Junior Engineer - Environment 18-02-2018 PDFCheng EngiNo ratings yet


















































































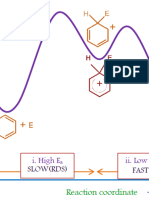



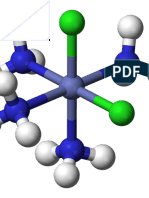





























Download as pdf or txt
You might also like
- Chem Lab Report - Electronic Absorption Spectra of Some Cu ComplexesDocument6 pagesChem Lab Report - Electronic Absorption Spectra of Some Cu ComplexesMiguel Ackah-Yensu93% (14)
- XRD Lab ReportDocument3 pagesXRD Lab ReportArman Boroomand67% (3)
- en 12004-2-2017 CenDocument35 pagesen 12004-2-2017 CenMalak Hindi100% (2)
- This Study Resource Was: SOILS 101 Chapter 5 AssignmentDocument5 pagesThis Study Resource Was: SOILS 101 Chapter 5 AssignmentSunil DhankharNo ratings yet
- Ligand Field StrengthDocument24 pagesLigand Field StrengthIrvandar NurviandyNo ratings yet
- CFT PDFDocument20 pagesCFT PDFRUFAS KANIKANTINo ratings yet
- CBSE Class 12 Chemistry Paper Sample Paper Solution Set 4Document14 pagesCBSE Class 12 Chemistry Paper Sample Paper Solution Set 4Gamer ChannelNo ratings yet
- Atomic Structure (1-35)Document35 pagesAtomic Structure (1-35)deepakkr08075% (4)
- Optical Properties of Metal Nanoparticles: Sriharsha KarumuriDocument12 pagesOptical Properties of Metal Nanoparticles: Sriharsha Karumurigea1616No ratings yet
- Interpretation of The Emission Spectra of Trivalent Chromium-Doped Garnet Crystals Using Tanabe-Sugano DiagramsDocument3 pagesInterpretation of The Emission Spectra of Trivalent Chromium-Doped Garnet Crystals Using Tanabe-Sugano DiagramsDaniela Araújo RodríguezNo ratings yet
- Transition Metal 4Document4 pagesTransition Metal 4Sushant ShahNo ratings yet
- Chapter 3Document47 pagesChapter 3蘇翊愷No ratings yet
- Monolayer-Protected Metal Nanoparticle: Toluene) Auc L Toluene) N (C H Auc L N (C HDocument27 pagesMonolayer-Protected Metal Nanoparticle: Toluene) Auc L Toluene) N (C H Auc L N (C HmichsantosNo ratings yet
- Color of Transition Metal IonsDocument19 pagesColor of Transition Metal IonsSyedah Maira ShahNo ratings yet
- (13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsDocument10 pages(13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsVikash KumarNo ratings yet
- Structure of Atom Class 11 Chemistry CBSE BoardDocument5 pagesStructure of Atom Class 11 Chemistry CBSE BoardminimataNo ratings yet
- Structure of Atom: Sub-Atomic Particles: Name Symbol Charge/C Relative Charge Mass/kgDocument8 pagesStructure of Atom: Sub-Atomic Particles: Name Symbol Charge/C Relative Charge Mass/kgSparsh MehtaNo ratings yet
- Solid State ChemistryDocument36 pagesSolid State ChemistrySoumya BullaNo ratings yet
- Chapter 2 - Structure of AtomDocument18 pagesChapter 2 - Structure of AtomstudyforiittomeetbtsNo ratings yet
- I. 1. What Are Nanoparticles?: Figure 1. Dimension of Different Objects in The Length ScaleDocument11 pagesI. 1. What Are Nanoparticles?: Figure 1. Dimension of Different Objects in The Length ScaleAmandeep Singh PannuNo ratings yet
- S/Sciences/Physics/Optics/Lasertut Orial/Creating/Creating - HTMDocument13 pagesS/Sciences/Physics/Optics/Lasertut Orial/Creating/Creating - HTMSuvankar ChakrabortyNo ratings yet
- CH 2 Structure of Atom PDFDocument18 pagesCH 2 Structure of Atom PDFjazi_4uNo ratings yet
- Grain Boundaries in MetalsDocument2 pagesGrain Boundaries in MetalsAvanish KumarNo ratings yet
- Atomic StructureDocument10 pagesAtomic StructureSatyam MittalNo ratings yet
- D and F BlockDocument8 pagesD and F BlockAnanyaNo ratings yet
- Lecture 2 CY101Document21 pagesLecture 2 CY101Gyanaranjan SahooNo ratings yet
- 11 Chemistry Notes Ch02 Structure of AtomDocument18 pages11 Chemistry Notes Ch02 Structure of Atomyuvrajbrand01No ratings yet
- Teori Meda KristalDocument41 pagesTeori Meda KristalGilang Maulana PutraNo ratings yet
- 2008 Tuning Luminescence Intensity of RHO6G Dye Using SilverDocument4 pages2008 Tuning Luminescence Intensity of RHO6G Dye Using Silverantoich antoichNo ratings yet
- Bonding Models in Inorganic Chemistry 1. Ionic CompoundsDocument12 pagesBonding Models in Inorganic Chemistry 1. Ionic Compoundsfavourabiola123No ratings yet
- 11-CHEMISTRY (CH-2) PART-1Document3 pages11-CHEMISTRY (CH-2) PART-1mt3835570No ratings yet
- Structure of AtomDocument72 pagesStructure of AtomAditi YadavNo ratings yet
- Chapter 12 - Atoms-Saju-Hsslive PDFDocument9 pagesChapter 12 - Atoms-Saju-Hsslive PDFAmiNo ratings yet
- Raman Scattering: Jordan University of Science and TechnologyDocument40 pagesRaman Scattering: Jordan University of Science and TechnologyhaimantiNo ratings yet
- Crystal Field Theory - NURDocument5 pagesCrystal Field Theory - NURNurhajrahNo ratings yet
- Initial Oxidation Rate of Metals and The Logarithmic EquationDocument14 pagesInitial Oxidation Rate of Metals and The Logarithmic EquationAndrea CalderaNo ratings yet
- 11 Chemistry Notes Ch02 Structure of AtomDocument18 pages11 Chemistry Notes Ch02 Structure of AtomSayantanBanerjee0% (1)
- Co Ordination 1Document33 pagesCo Ordination 1Nivi RajNo ratings yet
- Physicsatoms 46198Document11 pagesPhysicsatoms 46198user 003No ratings yet
- Atomic Structure PDFDocument40 pagesAtomic Structure PDFAnkit MaanNo ratings yet
- Chemistry Form 6 Sem 2 03Document45 pagesChemistry Form 6 Sem 2 03Ng Swee Loong StevenNo ratings yet
- Structure of Atom - 404Document72 pagesStructure of Atom - 404vipulvidhya2020No ratings yet
- Structure of AtomDocument8 pagesStructure of AtompreetikoravNo ratings yet
- CHM 112Document16 pagesCHM 112olamideolafimihan040No ratings yet
- Permittivity and Transmission of MetalsDocument3 pagesPermittivity and Transmission of MetalsmaxNo ratings yet
- Iit - JEE Syllabus: RSM79 PH I AS CH 1Document48 pagesIit - JEE Syllabus: RSM79 PH I AS CH 1MD IMRAN100% (1)
- 2.structure of Atom-Smart Booklet-1Document44 pages2.structure of Atom-Smart Booklet-1nadeemnagthan008No ratings yet
- Inorganic Chem 1 2 PDFDocument73 pagesInorganic Chem 1 2 PDFYT ChongNo ratings yet
- PNGE352 - Lec3 - GR Logs-April1a - 2022Document22 pagesPNGE352 - Lec3 - GR Logs-April1a - 2022adnan.bunyNo ratings yet
- Structure of Atom Class 11Document74 pagesStructure of Atom Class 11Komal VermaNo ratings yet
- Energy Level SplittingDocument4 pagesEnergy Level SplittingMa'arif A. SyafiiNo ratings yet
- DFFHMDocument9 pagesDFFHMyaswanthNo ratings yet
- Atoms and NucleiDocument66 pagesAtoms and NucleiphysicsbypaulsirNo ratings yet
- Atomic and Nuclear Physics: Thomson's Atomic ModelDocument23 pagesAtomic and Nuclear Physics: Thomson's Atomic ModelDHINESH HARIKUMARNo ratings yet
- Atoms:: Particle Electron Proton Neutron Discovery Nature of Charge Negative Amount of Charge MassDocument6 pagesAtoms:: Particle Electron Proton Neutron Discovery Nature of Charge Negative Amount of Charge MassNasser SsennogaNo ratings yet
- Atomic NucleusDocument15 pagesAtomic Nucleussreenivas1990100% (1)
- Amorphous Semiconductors: Structural, Optical, and Electronic PropertiesFrom EverandAmorphous Semiconductors: Structural, Optical, and Electronic PropertiesNo ratings yet
- Mechanism of Organic ReactionDocument4 pagesMechanism of Organic ReactionRSLNo ratings yet
- Colloidal Gold. Part I: Historical and Preparative Aspects, Morphology and StructureDocument6 pagesColloidal Gold. Part I: Historical and Preparative Aspects, Morphology and StructureRSL100% (1)
- Carbon Monoxide or Carbonyl: MO DescriptionDocument3 pagesCarbon Monoxide or Carbonyl: MO DescriptionRSLNo ratings yet
- NH4BH4Document1 pageNH4BH4RSLNo ratings yet
- Coordination Isomers ListDocument1 pageCoordination Isomers ListRSLNo ratings yet
- Basics of ElectrochemistryDocument22 pagesBasics of ElectrochemistryRSLNo ratings yet
- DebereinerDocument4 pagesDebereinerRSLNo ratings yet
- S-Block Elments: Inorganic ChemistryDocument8 pagesS-Block Elments: Inorganic ChemistryRSLNo ratings yet
- Epoxides Ring-Opening - Chemistry LibreTextsDocument3 pagesEpoxides Ring-Opening - Chemistry LibreTextsRSLNo ratings yet
- © 1934 Nature Publishing GroupDocument2 pages© 1934 Nature Publishing GroupRSLNo ratings yet
- Colloids: Thomas Graham (1861) Studied The Ability of Dissolved Substances ToDocument28 pagesColloids: Thomas Graham (1861) Studied The Ability of Dissolved Substances ToRSLNo ratings yet
- List of Straight-Chain AlkanesDocument6 pagesList of Straight-Chain AlkanesRSLNo ratings yet
- Piezoelectric Materials: Crystal Orientation and Poling Direction - COMSOL Blog PDFDocument4 pagesPiezoelectric Materials: Crystal Orientation and Poling Direction - COMSOL Blog PDFRSLNo ratings yet
- Priority List IUPACDocument1 pagePriority List IUPACRSLNo ratings yet
- 2019dec-03 - Ionic Equilibrium - PracticeSheetDocument2 pages2019dec-03 - Ionic Equilibrium - PracticeSheetRSLNo ratings yet
- Redox Reactions and Balancing Using Oxidation Number & NfactorDocument1 pageRedox Reactions and Balancing Using Oxidation Number & NfactorRSLNo ratings yet
- Analysis Report of Criminal and Financial Background Details of Winners in Lok Sabha 2019 ElectionsDocument397 pagesAnalysis Report of Criminal and Financial Background Details of Winners in Lok Sabha 2019 ElectionsRSLNo ratings yet
- Colligative Properties: Cryoscopy & EbulliosDocument30 pagesColligative Properties: Cryoscopy & EbulliosRSL100% (1)
- Ionic Equilibrium - Salt Hydrolysis Practice Questions (Level-1)Document3 pagesIonic Equilibrium - Salt Hydrolysis Practice Questions (Level-1)RSLNo ratings yet
- Isoelectric Point, PiDocument2 pagesIsoelectric Point, PiRSLNo ratings yet
- EAS Worksheet 1 SubjDocument1 pageEAS Worksheet 1 SubjRSLNo ratings yet
- Koopmans TheoremDocument14 pagesKoopmans TheoremRSLNo ratings yet
- The World of Chemistry Episode 5 - A Matter of StateDocument2 pagesThe World of Chemistry Episode 5 - A Matter of StateRSL0% (1)
- Chemistry Organic GOC Reaction Mechanism Elimination E1 E2 E1cBDocument4 pagesChemistry Organic GOC Reaction Mechanism Elimination E1 E2 E1cBRSL100% (1)
- Crossword Coordination CompoundsDocument2 pagesCrossword Coordination CompoundsRSLNo ratings yet
- Factors Affecting Relative Strengths of Acids and BasesDocument1 pageFactors Affecting Relative Strengths of Acids and BasesRSL80% (5)
- Fibre Epoxy Composites at Low Temperature HartwigDocument9 pagesFibre Epoxy Composites at Low Temperature HartwigodormicchiNo ratings yet
- Handbook of Advanced Plasma Processing Techniques Book ReviewDocument1 pageHandbook of Advanced Plasma Processing Techniques Book Reviewmustafa alasadyNo ratings yet
- Proofex GPXDocument2 pagesProofex GPXVenkata RaoNo ratings yet
- Rocky 3 Technical ManualDocument8 pagesRocky 3 Technical ManualMáximo ValdésNo ratings yet
- Measuring Adhesion by Tape Test: Standard Test Methods ForDocument7 pagesMeasuring Adhesion by Tape Test: Standard Test Methods ForJony NavaNo ratings yet
- CE 321 Chapter 7 Express For PDFDocument43 pagesCE 321 Chapter 7 Express For PDFGerald MoaresNo ratings yet
- Principles of AdhesionDocument44 pagesPrinciples of AdhesionOmar Hesham SalahNo ratings yet
- NRCC 53261Document18 pagesNRCC 53261MEHDI FARROKHINo ratings yet
- An Alternative Method For Thermal Cycling Test - Effect On The Marginal Microleakage and Bond Strength of Dental Polymer Bonded To Dentin PDFDocument5 pagesAn Alternative Method For Thermal Cycling Test - Effect On The Marginal Microleakage and Bond Strength of Dental Polymer Bonded To Dentin PDFilich sevillaNo ratings yet
- Kinetic Molecular Model of Solids and Liquids Activity 1: Color DropDocument12 pagesKinetic Molecular Model of Solids and Liquids Activity 1: Color DropElaine Mae G. EsqueroNo ratings yet
- Concepts of Surface TensionDocument31 pagesConcepts of Surface TensionMartin AdriazolaNo ratings yet
- BS 3900 Pull Off Test PDFDocument9 pagesBS 3900 Pull Off Test PDFAvinash LalNo ratings yet
- Particle Size Effect On The Hydrophobicity and The Natural Floatability of MolybdeniteDocument105 pagesParticle Size Effect On The Hydrophobicity and The Natural Floatability of MolybdeniteAgustín F. Correa100% (1)
- Bitumen Binder and TypesDocument47 pagesBitumen Binder and TypesTarun Goel50% (2)
- Keur 00824-401 201505Document4 pagesKeur 00824-401 201505erik0007No ratings yet
- Proofex 12 PDFDocument2 pagesProofex 12 PDFmilanbrasinaNo ratings yet
- 2023 - Gordon - Surface Enineering Polymeric Materials Towards Lunar Dust MitigationDocument6 pages2023 - Gordon - Surface Enineering Polymeric Materials Towards Lunar Dust Mitigationjames.millsNo ratings yet
- Contact Angles: Surface TensionDocument4 pagesContact Angles: Surface TensionArpan GhoshNo ratings yet
- 1 Corrosion Prevention by Protective Coatings (Munger)Document19 pages1 Corrosion Prevention by Protective Coatings (Munger)Gagan MehandirattaNo ratings yet
- A006 Pi VtmoeoDocument3 pagesA006 Pi VtmoeoRick LeeKang WangNo ratings yet
- D 1779 - 98 R04 Rde3nzk - PDFDocument3 pagesD 1779 - 98 R04 Rde3nzk - PDFMarceloNo ratings yet
- Astm D7234-19Document9 pagesAstm D7234-19AhmedNo ratings yet
- The Potential of Using Agricultural Waste: Corn Husk For Particleboard Raw MaterialDocument9 pagesThe Potential of Using Agricultural Waste: Corn Husk For Particleboard Raw MaterialDessy BelmonteNo ratings yet
- Inks For Printing On Plastic FilmsDocument8 pagesInks For Printing On Plastic FilmsLucas Del PretteNo ratings yet
- ASTM D429 Rubber To Metal Adhesion Test EquipmentDocument4 pagesASTM D429 Rubber To Metal Adhesion Test EquipmentLimNo ratings yet
- Waste Plastic Modified BituminDocument11 pagesWaste Plastic Modified BituminMd.imthiyazNo ratings yet
- Plasmatreat ImagebrochureDocument28 pagesPlasmatreat ImagebrochurechibigarNo ratings yet
- GSECL Vidyut Sahayak Junior Engineer - Environment 18-02-2018 PDFDocument20 pagesGSECL Vidyut Sahayak Junior Engineer - Environment 18-02-2018 PDFCheng EngiNo ratings yet