Download as pdf or txt
You might also like
- Digestion and Absorption in The Gastrointestinal Tract-Summary of GuytonDocument7 pagesDigestion and Absorption in The Gastrointestinal Tract-Summary of GuytonLeira Lee100% (1)
- Biology For CAPE Unit 2 Chapter 2 AnswersDocument9 pagesBiology For CAPE Unit 2 Chapter 2 AnswersFiveLimaRomeo83% (6)
- Digestion and Absorption of Lipid 12Document36 pagesDigestion and Absorption of Lipid 12Pranjul MishraNo ratings yet
- Card 3 Digestion Absorption Metabolism 5512Document210 pagesCard 3 Digestion Absorption Metabolism 5512Suraiya IslamNo ratings yet
- BIOCHEM DigestionDocument53 pagesBIOCHEM DigestionAngelika Perez CunanNo ratings yet
- Kuliah II - Digestive KH, Lemak, Phosphat Asma NukleatDocument30 pagesKuliah II - Digestive KH, Lemak, Phosphat Asma NukleatHastomo Nur HidayatullohNo ratings yet
- Digestion SlideDocument18 pagesDigestion SlideezebelluciNo ratings yet
- Lipids Digestion and AbsorptionDocument28 pagesLipids Digestion and AbsorptionEng Matti Ur RehmanNo ratings yet
- Week 14: Digestion Chemistry: Intrinsic FactorDocument4 pagesWeek 14: Digestion Chemistry: Intrinsic FactorLore Anne Mhae SantosNo ratings yet
- Salivary SecretionDocument10 pagesSalivary SecretionMahadi Hasan KhanNo ratings yet
- The Biochemistry of Digestion, Absorption and Detoxification by Prof. Dr. Hedef D. El-YassinDocument64 pagesThe Biochemistry of Digestion, Absorption and Detoxification by Prof. Dr. Hedef D. El-YassinSayhidoen CepexNo ratings yet
- Expt 10 DigestionDocument10 pagesExpt 10 DigestionRuby Sarai BelinoNo ratings yet
- K4 - Biokimia GIDocument42 pagesK4 - Biokimia GIsilakan_isiNo ratings yet
- Digestive GlandDocument22 pagesDigestive GlandMalinus TorquatusNo ratings yet
- Secretion of Bile and The Role of Bile Acids in DigestionDocument8 pagesSecretion of Bile and The Role of Bile Acids in DigestionMarianne Kristelle NonanNo ratings yet
- Lipid MetabolismDocument52 pagesLipid MetabolismDr.P.NatarajanNo ratings yet
- Prepared By: Richa KhatiwadaDocument10 pagesPrepared By: Richa KhatiwadaSabiha TabassumNo ratings yet
- Lipid Absorption Metabolism PPTX ModifiedDocument81 pagesLipid Absorption Metabolism PPTX Modifiedkel GetanehNo ratings yet
- Digestion: By: Roselita O. Natividad and Teresita G Montaño, PHD Nat. Science - Ateneo de Zamboanga UniversityDocument5 pagesDigestion: By: Roselita O. Natividad and Teresita G Montaño, PHD Nat. Science - Ateneo de Zamboanga UniversityMini BossNo ratings yet
- Digestion of Fats (Triglycerides/Lipids) : By: Brittany Speer, William Mcnees, Peter Schulz, Maiko Ibay, and Regina TabiDocument31 pagesDigestion of Fats (Triglycerides/Lipids) : By: Brittany Speer, William Mcnees, Peter Schulz, Maiko Ibay, and Regina Tabikhaer udinNo ratings yet
- DigestionDocument21 pagesDigestionAndersonNo ratings yet
- Sistem Pencernaan (Digestion System)Document28 pagesSistem Pencernaan (Digestion System)PaRaDoX eLaKSaMaNaNo ratings yet
- Carbohydrate, Protein and Lipid Metabolism-B.NDocument77 pagesCarbohydrate, Protein and Lipid Metabolism-B.NBusy worldNo ratings yet
- Biochemistry of GitDocument148 pagesBiochemistry of GitMedical NotesNo ratings yet
- Ch15 Metabolism of Dietary LipidsDocument37 pagesCh15 Metabolism of Dietary LipidsWahab KhaniNo ratings yet
- Carbohydrate, Lipid and Protein EssayDocument2 pagesCarbohydrate, Lipid and Protein EssayYEO MING HUI MoeNo ratings yet
- BiochemDocument10 pagesBiochemWaseem Abbas ShaikhNo ratings yet
- Biochemical Aspects of Digestion of Lipids: Dr. Amr S. MoustafaDocument30 pagesBiochemical Aspects of Digestion of Lipids: Dr. Amr S. Moustafaraanja2No ratings yet
- Compiled MetabolismDocument155 pagesCompiled MetabolismIced WatermelonNo ratings yet
- Digesti & Absorpsi MakananDocument109 pagesDigesti & Absorpsi MakananIntan NamiraNo ratings yet
- Session 6 Digestion Absorbtion Transport and Excretion of Nutrients PDFDocument63 pagesSession 6 Digestion Absorbtion Transport and Excretion of Nutrients PDFAbubakar SuleimanNo ratings yet
- Gastro Section CDocument11 pagesGastro Section CNadja JamilahNo ratings yet
- GruczołyDocument33 pagesGruczołynatalie muyengwaNo ratings yet
- Digestive SystemDocument33 pagesDigestive SystemSatyabrat DuttaNo ratings yet
- Wa0009.Document29 pagesWa0009.অণ্যরকম BDNo ratings yet
- Digestive System: Digestion and Absorption of Carbohydrates, Proteins and FatsDocument45 pagesDigestive System: Digestion and Absorption of Carbohydrates, Proteins and FatshiNo ratings yet
- Digestion & AbsorptionDocument18 pagesDigestion & AbsorptionDoghouseNo ratings yet
- FA MetabolismDocument23 pagesFA MetabolismMaham KhanNo ratings yet
- Approval SheetDocument24 pagesApproval SheetIntan AnggrainiNo ratings yet
- Case PresentationDocument15 pagesCase PresentationMarianne Del Rosario AndayaNo ratings yet
- Gallbladder & Bile Physiological AspectsDocument48 pagesGallbladder & Bile Physiological AspectsPhysiology by Dr RaghuveerNo ratings yet
- Biology Notes Human Digestive System For SSC Exam PDFDocument2 pagesBiology Notes Human Digestive System For SSC Exam PDFनितीन लाठकरNo ratings yet
- 2nd Sem PANCREAS, LIVER, BILEDocument34 pages2nd Sem PANCREAS, LIVER, BILEsanullah123khan.13No ratings yet
- Assignment Mammalian Physiology: Topic:-Mechanism of Digestion ForDocument6 pagesAssignment Mammalian Physiology: Topic:-Mechanism of Digestion ForRajkamal SarmaNo ratings yet
- Other Foreign Substances These Substances Have Entered The Blood Supply EitherDocument15 pagesOther Foreign Substances These Substances Have Entered The Blood Supply EitherAnis Assila Mohd RohaimiNo ratings yet
- Metabolic Functions of The LiverDocument3 pagesMetabolic Functions of The LiverAizat KamalNo ratings yet
- Week 6 LipidsDocument59 pagesWeek 6 Lipidsbo aNo ratings yet
- Digestion NoteDocument5 pagesDigestion Notebob burgersNo ratings yet
- Gastrointestinal Tract Function and DisordersDocument59 pagesGastrointestinal Tract Function and DisordersTaylorNo ratings yet
- Mouth: Lingual Lipase LipidDocument9 pagesMouth: Lingual Lipase LipidOscar Villados Jr.No ratings yet
- Bio Notes: Digestion (IB HL 6.1)Document6 pagesBio Notes: Digestion (IB HL 6.1)BekNo ratings yet
- git pqDocument18 pagesgit pqtaiwodamola789No ratings yet
- مرتضى 2Document11 pagesمرتضى 2mmalaui44No ratings yet
- Lipid Metabolism (I) - Lipid Digestion and AbsorptionDocument18 pagesLipid Metabolism (I) - Lipid Digestion and Absorptionlightning proNo ratings yet
- Digestive SystemDocument9 pagesDigestive SystemJhomar B, Dela CruzNo ratings yet
- Digestion and AbsorptionDocument6 pagesDigestion and AbsorptionE.AshwinNo ratings yet
- Lipids NotesDocument4 pagesLipids NoteskatNo ratings yet
- Digestion and Absorption - 101328Document10 pagesDigestion and Absorption - 101328Emmanuel IshiomaNo ratings yet
- Digestion: Proteins Fats CarbohydratesDocument42 pagesDigestion: Proteins Fats CarbohydratesVal Kay HeikeNo ratings yet
- Digestion and Absorption of CarbohydratesDocument28 pagesDigestion and Absorption of Carbohydratesshakila786No ratings yet
- Plant and Animal Bio-Chemistry - Including Information on Amino Acids, Proteins, Pigments and Other Chemical Constituents of Organic MatterFrom EverandPlant and Animal Bio-Chemistry - Including Information on Amino Acids, Proteins, Pigments and Other Chemical Constituents of Organic MatterNo ratings yet
- KSD Odhfowi 4 FH'Document5 pagesKSD Odhfowi 4 FH'Alondra SagarioNo ratings yet
- Jkxcshweiufg WDocument1 pageJkxcshweiufg WAlondra SagarioNo ratings yet
- KA LEHFIO WHFKJWDocument15 pagesKA LEHFIO WHFKJWAlondra SagarioNo ratings yet
- ELECTROLYTES (Na & K)Document3 pagesELECTROLYTES (Na & K)Alondra SagarioNo ratings yet
- ABO DiscrepanciesDocument40 pagesABO DiscrepanciesAlondra Sagario100% (1)
- Endocrinology Part 2Document4 pagesEndocrinology Part 2Alondra SagarioNo ratings yet
- Planning Health Education Programme 2Document9 pagesPlanning Health Education Programme 2Alondra SagarioNo ratings yet
- Is QuestionsDocument2 pagesIs QuestionsAlondra SagarioNo ratings yet
- Rights and Responsibilities of Health Care ProvidersDocument2 pagesRights and Responsibilities of Health Care ProvidersAlondra Sagario0% (1)
- Anticoagulant of The Tube Sodium Citrate (Blue) Anticoagulant To Blood Ratio: 1:9 Oral Anticoagulant Therapy: WarfarinDocument2 pagesAnticoagulant of The Tube Sodium Citrate (Blue) Anticoagulant To Blood Ratio: 1:9 Oral Anticoagulant Therapy: WarfarinAlondra SagarioNo ratings yet
- Demonstrating The Nature and Function of Biomolecules: CarbohydratesDocument50 pagesDemonstrating The Nature and Function of Biomolecules: CarbohydratesAlondra SagarioNo ratings yet
- Phce2572804417664 1Document1 pagePhce2572804417664 1Alondra SagarioNo ratings yet
- ThrombolysisDocument13 pagesThrombolysisAlondra SagarioNo ratings yet
- Structure Versus Consideration Leadership StylesDocument3 pagesStructure Versus Consideration Leadership StylesAlondra SagarioNo ratings yet
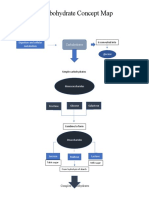
- Carbohydrate Concept Map: Digestion and Cellular Metabolism GlucoseDocument2 pagesCarbohydrate Concept Map: Digestion and Cellular Metabolism GlucoseAlondra SagarioNo ratings yet
- 3.8 Matunog-Act 3.8 Biocm1 June 2020Document2 pages3.8 Matunog-Act 3.8 Biocm1 June 2020Alondra SagarioNo ratings yet
- Alondra Sagario A.Document3 pagesAlondra Sagario A.Alondra SagarioNo ratings yet
- Functions of ArtDocument2 pagesFunctions of ArtAlondra SagarioNo ratings yet
- 5 Management of ChangeDocument3 pages5 Management of ChangeAlondra SagarioNo ratings yet
- LABMAN 1-2.1: Management ProcessDocument5 pagesLABMAN 1-2.1: Management ProcessAlondra SagarioNo ratings yet
- Standby Lecture Starts: Coverage: Financial and Technical ManagementDocument32 pagesStandby Lecture Starts: Coverage: Financial and Technical ManagementAlondra SagarioNo ratings yet
- 3.8 Matunog-Act 3.8 Biocm1 June 2020Document2 pages3.8 Matunog-Act 3.8 Biocm1 June 2020Alondra SagarioNo ratings yet
- LAbMan 2.2 Reviewer 1Document2 pagesLAbMan 2.2 Reviewer 1Alondra SagarioNo ratings yet
- Water Quality Assessment of Carangan Estero in Ozamiz City, PhilippinesDocument26 pagesWater Quality Assessment of Carangan Estero in Ozamiz City, PhilippinesAlondra SagarioNo ratings yet
- 3.5 Enzymes 2Document64 pages3.5 Enzymes 2Alondra SagarioNo ratings yet
- Biochem - Vit.B12 Role in MetabolismDocument3 pagesBiochem - Vit.B12 Role in MetabolismAneeza Ahmad100% (1)
- Final BiochemistryDocument6 pagesFinal BiochemistryHuyen Tram NguyenNo ratings yet
- Digestive and Respiratory SystemDocument48 pagesDigestive and Respiratory Systemkenneth vergaraNo ratings yet
- MC2 Lec11 Catabolic PathwaysDocument61 pagesMC2 Lec11 Catabolic PathwaysLauren CarlosIINo ratings yet
- 9.2 Packet Heaven WalkerDocument5 pages9.2 Packet Heaven WalkerHNo ratings yet
- Photosynthesis and RespirationDocument4 pagesPhotosynthesis and RespirationChristian BaleNo ratings yet
- Degradasi Karbohidrat: Drh.M.Isa, M.SiDocument32 pagesDegradasi Karbohidrat: Drh.M.Isa, M.SiReIzq RamaDhan100% (1)
- 2012 DSE Bio 1BDocument12 pages2012 DSE Bio 1BLeung Man ChunNo ratings yet
- Nutritional Management of Chronic KidneyDocument12 pagesNutritional Management of Chronic KidneyMARIA CASTELLANOSNo ratings yet
- Glycolysis - LippincottsDocument11 pagesGlycolysis - LippincottsJulia QuimadaNo ratings yet
- Harold Baum The Biochemists Songbook CRC 2004Document107 pagesHarold Baum The Biochemists Songbook CRC 2004Nguyen Bao Son Truong THPT chuyen Hoang Le KhaNo ratings yet
- Specific Catabolic Pathways: Carbohydrate, Lipid, and Protein MetabolismDocument43 pagesSpecific Catabolic Pathways: Carbohydrate, Lipid, and Protein MetabolismShereen AlobinayNo ratings yet
- Anatomy & Plant PhysiologyDocument27 pagesAnatomy & Plant Physiologyshree devNo ratings yet
- BIOCHEMISTRY BOARD EXAM QUESTIONS - QuestionDocument7 pagesBIOCHEMISTRY BOARD EXAM QUESTIONS - QuestionchristinejoanNo ratings yet
- Metabolism of CHOsDocument32 pagesMetabolism of CHOsRajinder ChawlaNo ratings yet
- Glukoneogenesis: Prof - Dr. Suhartati, DR., MSDocument25 pagesGlukoneogenesis: Prof - Dr. Suhartati, DR., MSDinda DhitaNo ratings yet
- Food Science Nutrition - 2019 - Yang - Citric Acid Treatment Reduces Decay and Maintains The Postharvest Quality of PeachDocument9 pagesFood Science Nutrition - 2019 - Yang - Citric Acid Treatment Reduces Decay and Maintains The Postharvest Quality of PeachTeena TutorNo ratings yet
- Ketogenic Diet For Human HealthDocument4 pagesKetogenic Diet For Human Healthمحمد عبدالرحيم هلالNo ratings yet
- Carbohydrate MetabolismDocument8 pagesCarbohydrate MetabolismChaudhari Saroj100% (1)
- Piperine For ObesityDocument8 pagesPiperine For ObesityTalluri GaneshNo ratings yet
- Wardlaws Perspectives in Nutrition A Functional Approach 1st Edition Byrd Bredbenner Test BankDocument38 pagesWardlaws Perspectives in Nutrition A Functional Approach 1st Edition Byrd Bredbenner Test Bankedricduyenuc1uw100% (26)
- Bio 93 Midterm 2 Review Session KEYDocument121 pagesBio 93 Midterm 2 Review Session KEYKenosNo ratings yet
- Ecoli Citrate Metabolism Evolution - STDDocument81 pagesEcoli Citrate Metabolism Evolution - STDDerrick BuchananNo ratings yet
- Unit 4 BiologyDocument9 pagesUnit 4 BiologyChristine MoniqueNo ratings yet
- How Cells Harvest EnergyDocument26 pagesHow Cells Harvest EnergyMarita YaghiNo ratings yet
- Bichem Module - 1-2014 With 1 and 2Document41 pagesBichem Module - 1-2014 With 1 and 2Mukunda MurariNo ratings yet
- Review: Mitochondria: in Sickness and in HealthDocument15 pagesReview: Mitochondria: in Sickness and in HealthPetra JobovaNo ratings yet
- Chapter 19 Citric Acide CycleDocument37 pagesChapter 19 Citric Acide CycleWaad MajidNo ratings yet
- Instant Download PDF Wardlaws Perspectives in Nutrition A Functional Approach 1st Edition Byrd-Bredbenner Test Bank Full ChapterDocument60 pagesInstant Download PDF Wardlaws Perspectives in Nutrition A Functional Approach 1st Edition Byrd-Bredbenner Test Bank Full Chapterpeneasarjan100% (3)