
CRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular Biology
CRISPR/Cas9 & Targeted Genome Editing: New Era in Molecular Biology
You might also like
- Lab Report CRISPR Sample 1Document9 pagesLab Report CRISPR Sample 1mayagal1707No ratings yet
- Rewriting A GenomeDocument2 pagesRewriting A GenomeUjwalNo ratings yet
- Crispr Cas9 and Targeted Genome EditingDocument5 pagesCrispr Cas9 and Targeted Genome EditingDaniH46No ratings yet
- Editing Plant Genomes With Crispr/Cas9: SciencedirectDocument9 pagesEditing Plant Genomes With Crispr/Cas9: SciencedirectVictor BonillaNo ratings yet
- Lec 2 Gene Editing Tools Part 1Document23 pagesLec 2 Gene Editing Tools Part 1mahhmoNo ratings yet
- CRISPaint Allows Modular Base-Specific Gene TagginDocument12 pagesCRISPaint Allows Modular Base-Specific Gene TagginJyotirmayee TalapatraNo ratings yet
- The FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Document8 pagesThe FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Alberto Luis Lizcano GonzálezNo ratings yet
- CRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesDocument7 pagesCRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesAngelica RestrepoNo ratings yet
- BIO 254 Experiment - 2Document15 pagesBIO 254 Experiment - 2bursalibeyza494No ratings yet
- Miki2021 Protocol CRISPRCas9-BasedGenomeEditingTDocument26 pagesMiki2021 Protocol CRISPRCas9-BasedGenomeEditingTSergio D. PérezNo ratings yet
- Prof Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingDocument9 pagesProf Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingkonarkNo ratings yet
- Wolter & Putcha (2018) - Plant Transcription FactorsDocument18 pagesWolter & Putcha (2018) - Plant Transcription FactorsAna Luiza Atella de FreitasNo ratings yet
- Supplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Document36 pagesSupplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Ronilo Jose Danila FloresNo ratings yet
- Kon Erm Ann 2014Document18 pagesKon Erm Ann 2014Letícia AlibertiNo ratings yet
- Crispr Articulo XDXDXDTTTTTTTTTTTTTTTT PDFDocument9 pagesCrispr Articulo XDXDXDTTTTTTTTTTTTTTTT PDFDany NahomiNo ratings yet
- Clover KnockinDocument14 pagesClover Knockingemmen99No ratings yet
- Ma 2014Document20 pagesMa 2014hiwmacrigeeeNo ratings yet
- Protein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9Document13 pagesProtein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9MichaelandKaye DanaoNo ratings yet
- CRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsDocument14 pagesCRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsAli HamzaNo ratings yet
- Addgene CRISPR GuideDocument18 pagesAddgene CRISPR GuideThomasNo ratings yet
- Crispr Cas 9Document3 pagesCrispr Cas 9E narender nayakNo ratings yet
- CRISPR1Document7 pagesCRISPR1cesarNo ratings yet
- Infographic - CRISPR - Cas9Document3 pagesInfographic - CRISPR - Cas9felipe duarteNo ratings yet
- Crispr BMB402 2022Document36 pagesCrispr BMB402 2022Mohadev SahaNo ratings yet
- ReportDocument6 pagesReportramna ziaNo ratings yet
- Raymond Et Al 2018 General Method For Plasmid Construction Using Homologous RecombinationDocument6 pagesRaymond Et Al 2018 General Method For Plasmid Construction Using Homologous Recombinationdaniel.murphyagudoNo ratings yet
- Vakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage EventsDocument8 pagesVakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage Eventsירדן לויןNo ratings yet
- Gabr Mahmoud ThesisDocument35 pagesGabr Mahmoud Thesismahmoud gabrNo ratings yet
- Senturk Et Al. 2015Document10 pagesSenturk Et Al. 2015Georgina Meza RadillaNo ratings yet
- Cas 9Document28 pagesCas 9Rin ChanNo ratings yet
- CRISPR Cas9 TechniqueDocument9 pagesCRISPR Cas9 TechniqueAdil ZahoorNo ratings yet
- Crispr Cas9 PDFDocument16 pagesCrispr Cas9 PDFIves LucasNo ratings yet
- Applications of CRISPR-Cas For Synthetic Biology and Genetic RecordingDocument7 pagesApplications of CRISPR-Cas For Synthetic Biology and Genetic RecordingShampa SenNo ratings yet
- Crispr Week 12-13 by AyeshaDocument12 pagesCrispr Week 12-13 by AyeshaAyesha KhalidNo ratings yet
- Efficient CRISPR Editing With A Hypercompact Cas12Document14 pagesEfficient CRISPR Editing With A Hypercompact Cas12Aimen FatimaNo ratings yet
- Crispr Cas 9Document12 pagesCrispr Cas 9ritika0206kalraNo ratings yet
- Advances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceDocument13 pagesAdvances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceAmina Tucak-SmajićNo ratings yet
- ZFN, Talens, Crispr PDFDocument9 pagesZFN, Talens, Crispr PDFjezelle lividNo ratings yet
- Synthetic Chimeric Nucleases Function For Ef Ficient Genome EditingDocument11 pagesSynthetic Chimeric Nucleases Function For Ef Ficient Genome EditingFeng YueNo ratings yet
- Jove 138 57560Document8 pagesJove 138 57560lauyehsiangNo ratings yet
- N Comms 13274Document7 pagesN Comms 13274doniNo ratings yet
- 10 1016@j TRSL 2015 09 008Document17 pages10 1016@j TRSL 2015 09 008hiwmacrigeeeNo ratings yet
- Research Problem Assignment Week 6Document3 pagesResearch Problem Assignment Week 6Pearlyn LauNo ratings yet
- Crispr Cas 1Document10 pagesCrispr Cas 1ABHISHEK SWARNAKARNo ratings yet
- Gene Editing Prime Time 10.1056@NEJMcibr1914271Document4 pagesGene Editing Prime Time 10.1056@NEJMcibr1914271MCuk2606No ratings yet
- CRISPR - Nov27 - 2023 - TaggedDocument55 pagesCRISPR - Nov27 - 2023 - TaggedRitwik DasNo ratings yet
- 2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemDocument12 pages2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemCristian Felipe Sandoval QuiñonezNo ratings yet
- 3 Gene Editing DocumentDocument4 pages3 Gene Editing DocumentAnanya ChauhanNo ratings yet
- CRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumDocument11 pagesCRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumIram AmmadNo ratings yet
- Targeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesDocument21 pagesTargeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesSabranth GuptaNo ratings yet
- Genome EditingDocument12 pagesGenome EditingDahlia StudioNo ratings yet
- NEB Featurearticle Crispr Cas9 Summer2014 LowresDocument6 pagesNEB Featurearticle Crispr Cas9 Summer2014 LowresRakhee RachelNo ratings yet
- A Protocol For Designing Sirnas With High Functionality and SpecificityDocument11 pagesA Protocol For Designing Sirnas With High Functionality and SpecificityThoha AlbaarNo ratings yet
- CRISPR Activation and Interference As Investigative ToolsDocument8 pagesCRISPR Activation and Interference As Investigative ToolsannyoryaNo ratings yet
- Genome Editing and Crispr Cas 9Document12 pagesGenome Editing and Crispr Cas 9HemsaiYadavNo ratings yet
- CRISPRDocument15 pagesCRISPRanirbanmanna88320No ratings yet
- Nobel Prize in Chemistry 2020: Institute of Agriculture and Animal Sciences (Iaas)Document5 pagesNobel Prize in Chemistry 2020: Institute of Agriculture and Animal Sciences (Iaas)Ramesh JØshiNo ratings yet
- A Review On The Mechanism and Applications of CRISPR Cas9 Cas12 Cas13 Cas14 Proteins Utilized For Genome EngineeringDocument15 pagesA Review On The Mechanism and Applications of CRISPR Cas9 Cas12 Cas13 Cas14 Proteins Utilized For Genome EngineeringrmbichiNo ratings yet
- Ijms 25 00823Document21 pagesIjms 25 00823Maria ClaraNo ratings yet
- DNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensFrom EverandDNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensNo ratings yet
- IDT - The CRISPR Basics HandbookDocument48 pagesIDT - The CRISPR Basics Handbookthauwui86100% (1)
- Application of Genetics and Biotechnology For Improving Medicinal PlantsDocument21 pagesApplication of Genetics and Biotechnology For Improving Medicinal PlantsAracely FernandaNo ratings yet
- Synthetic BiologyDocument38 pagesSynthetic BiologyHarry DouglasNo ratings yet
- ZFNDocument20 pagesZFNSenthilkumar PalanisamyNo ratings yet
- CRISPR 101 Ebook PDFDocument29 pagesCRISPR 101 Ebook PDFMarco Antonio Diaz GonzalezNo ratings yet
- Prahaar 3.0 Science SummaryDocument35 pagesPrahaar 3.0 Science Summaryjeshwanth2305No ratings yet
- GENE EDITING. Final PDFDocument13 pagesGENE EDITING. Final PDFS.A. DeeptiNo ratings yet
- Genetically Modified OrganismsDocument7 pagesGenetically Modified OrganismsPrajwal PatilNo ratings yet
- Restriction EnzymesDocument15 pagesRestriction EnzymesCharisma MeromiNo ratings yet
- Marine Microalgae For Production of Biofuels and Chemicals: SciencedirectDocument10 pagesMarine Microalgae For Production of Biofuels and Chemicals: SciencedirectShampa SenNo ratings yet
- Caft ReportDocument26 pagesCaft Reportrushi tahakikNo ratings yet
- s12929 018 0425 5 PDFDocument18 pagess12929 018 0425 5 PDFSURYONONo ratings yet
- Splat BookDocument170 pagesSplat Booksunny kuttanNo ratings yet
- ZFN, Talens, Crispr PDFDocument9 pagesZFN, Talens, Crispr PDFjezelle lividNo ratings yet
- UC Opp No. 2 CRISPR PatentDocument65 pagesUC Opp No. 2 CRISPR PatentjsherkowNo ratings yet
- Recent Advances in Animal Nutrition and Metabolism, Guoyao WuDocument344 pagesRecent Advances in Animal Nutrition and Metabolism, Guoyao WuPatricia TorresNo ratings yet
- Genome Engineeering Using Crispr Cas 9 System: June 2019Document8 pagesGenome Engineeering Using Crispr Cas 9 System: June 2019Mihaela CrafcenkoNo ratings yet
- Precision Medicine, CRISPR, and Genome Engineering: Stephen H. Tsang EditorDocument180 pagesPrecision Medicine, CRISPR, and Genome Engineering: Stephen H. Tsang EditorVinay Sagar LNo ratings yet
- Genetic EngineeringDocument26 pagesGenetic EngineeringMichaelAbdonDomingoFavoNo ratings yet
- Advanced Therapeutics - 2021 - ShahryariDocument42 pagesAdvanced Therapeutics - 2021 - ShahryariShubham AnandNo ratings yet
- Bio ProjectDocument11 pagesBio ProjectKshitij SharmaNo ratings yet
- Genome Editing An Ethical ReviewDocument136 pagesGenome Editing An Ethical ReviewIvica KelamNo ratings yet
- CRISPR+101+eBook v2021Document20 pagesCRISPR+101+eBook v2021Ruan GonçalvesNo ratings yet
- EDICIÓN GENÉTICA CRISPRcasDocument50 pagesEDICIÓN GENÉTICA CRISPRcasMAKARENA JIMENEZ VILLEGASNo ratings yet
- Liver Disease in Alpha-1 Antitrypsin Deficiency: Current Approaches and Future DirectionsDocument10 pagesLiver Disease in Alpha-1 Antitrypsin Deficiency: Current Approaches and Future DirectionsEianni Patrice De JoseNo ratings yet
- Synthego Ebook Crispr 101 PDFDocument29 pagesSynthego Ebook Crispr 101 PDFRie R100% (1)
- 2023 - Utilization of Genome Editing For Livestock Resilience in Changing EnvironmentDocument7 pages2023 - Utilization of Genome Editing For Livestock Resilience in Changing Environmentsoltani59No ratings yet
- ZFN, TALEN, and CRISPR-Cas-based Methods For Genome EngineeringDocument9 pagesZFN, TALEN, and CRISPR-Cas-based Methods For Genome EngineeringRomina Tamara Gil RamirezNo ratings yet
- Genetic Engineering of Microalgae For Enhanced Biorefi - 2020 - BiotechnologyDocument13 pagesGenetic Engineering of Microalgae For Enhanced Biorefi - 2020 - BiotechnologyerosNo ratings yet
- Editing Plant Genomes With Crispr/Cas9: SciencedirectDocument9 pagesEditing Plant Genomes With Crispr/Cas9: SciencedirectAli HamzaNo ratings yet























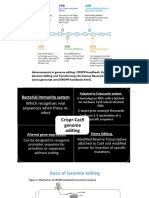
































































Download as docx, pdf, or txt
You might also like
- Lab Report CRISPR Sample 1Document9 pagesLab Report CRISPR Sample 1mayagal1707No ratings yet
- Rewriting A GenomeDocument2 pagesRewriting A GenomeUjwalNo ratings yet
- Crispr Cas9 and Targeted Genome EditingDocument5 pagesCrispr Cas9 and Targeted Genome EditingDaniH46No ratings yet
- Editing Plant Genomes With Crispr/Cas9: SciencedirectDocument9 pagesEditing Plant Genomes With Crispr/Cas9: SciencedirectVictor BonillaNo ratings yet
- Lec 2 Gene Editing Tools Part 1Document23 pagesLec 2 Gene Editing Tools Part 1mahhmoNo ratings yet
- CRISPaint Allows Modular Base-Specific Gene TagginDocument12 pagesCRISPaint Allows Modular Base-Specific Gene TagginJyotirmayee TalapatraNo ratings yet
- The FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Document8 pagesThe FEBS Journal - 2014 - Ma - Genome Modification by CRISPR Cas9Alberto Luis Lizcano GonzálezNo ratings yet
- CRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesDocument7 pagesCRISPR/Cas9 For Genome Editing: Progress, Implications and ChallengesAngelica RestrepoNo ratings yet
- BIO 254 Experiment - 2Document15 pagesBIO 254 Experiment - 2bursalibeyza494No ratings yet
- Miki2021 Protocol CRISPRCas9-BasedGenomeEditingTDocument26 pagesMiki2021 Protocol CRISPRCas9-BasedGenomeEditingTSergio D. PérezNo ratings yet
- Prof Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingDocument9 pagesProf Silberklang BTECH-480 Term Paper Topic-CRISPR Cas 9 Gene EditingkonarkNo ratings yet
- Wolter & Putcha (2018) - Plant Transcription FactorsDocument18 pagesWolter & Putcha (2018) - Plant Transcription FactorsAna Luiza Atella de FreitasNo ratings yet
- Supplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Document36 pagesSupplementary Materials For: RNA-Guided Human Genome Engineering Via Cas9Ronilo Jose Danila FloresNo ratings yet
- Kon Erm Ann 2014Document18 pagesKon Erm Ann 2014Letícia AlibertiNo ratings yet
- Crispr Articulo XDXDXDTTTTTTTTTTTTTTTT PDFDocument9 pagesCrispr Articulo XDXDXDTTTTTTTTTTTTTTTT PDFDany NahomiNo ratings yet
- Clover KnockinDocument14 pagesClover Knockingemmen99No ratings yet
- Ma 2014Document20 pagesMa 2014hiwmacrigeeeNo ratings yet
- Protein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9Document13 pagesProtein From Defluviimonas Sр.: Priority Application Title: DNA CUTTING MEANS BASED ON CAS9MichaelandKaye DanaoNo ratings yet
- CRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsDocument14 pagesCRISPR/Cas9 Platforms For Genome Editing in Plants: Developments and ApplicationsAli HamzaNo ratings yet
- Addgene CRISPR GuideDocument18 pagesAddgene CRISPR GuideThomasNo ratings yet
- Crispr Cas 9Document3 pagesCrispr Cas 9E narender nayakNo ratings yet
- CRISPR1Document7 pagesCRISPR1cesarNo ratings yet
- Infographic - CRISPR - Cas9Document3 pagesInfographic - CRISPR - Cas9felipe duarteNo ratings yet
- Crispr BMB402 2022Document36 pagesCrispr BMB402 2022Mohadev SahaNo ratings yet
- ReportDocument6 pagesReportramna ziaNo ratings yet
- Raymond Et Al 2018 General Method For Plasmid Construction Using Homologous RecombinationDocument6 pagesRaymond Et Al 2018 General Method For Plasmid Construction Using Homologous Recombinationdaniel.murphyagudoNo ratings yet
- Vakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage EventsDocument8 pagesVakulskas Behlke 2019 Evaluation and Reduction of Crispr Off Target Cleavage Eventsירדן לויןNo ratings yet
- Gabr Mahmoud ThesisDocument35 pagesGabr Mahmoud Thesismahmoud gabrNo ratings yet
- Senturk Et Al. 2015Document10 pagesSenturk Et Al. 2015Georgina Meza RadillaNo ratings yet
- Cas 9Document28 pagesCas 9Rin ChanNo ratings yet
- CRISPR Cas9 TechniqueDocument9 pagesCRISPR Cas9 TechniqueAdil ZahoorNo ratings yet
- Crispr Cas9 PDFDocument16 pagesCrispr Cas9 PDFIves LucasNo ratings yet
- Applications of CRISPR-Cas For Synthetic Biology and Genetic RecordingDocument7 pagesApplications of CRISPR-Cas For Synthetic Biology and Genetic RecordingShampa SenNo ratings yet
- Crispr Week 12-13 by AyeshaDocument12 pagesCrispr Week 12-13 by AyeshaAyesha KhalidNo ratings yet
- Efficient CRISPR Editing With A Hypercompact Cas12Document14 pagesEfficient CRISPR Editing With A Hypercompact Cas12Aimen FatimaNo ratings yet
- Crispr Cas 9Document12 pagesCrispr Cas 9ritika0206kalraNo ratings yet
- Advances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceDocument13 pagesAdvances in CRISPR-Cas9 Genome Engineering: Lessons Learned From RNA InterferenceAmina Tucak-SmajićNo ratings yet
- ZFN, Talens, Crispr PDFDocument9 pagesZFN, Talens, Crispr PDFjezelle lividNo ratings yet
- Synthetic Chimeric Nucleases Function For Ef Ficient Genome EditingDocument11 pagesSynthetic Chimeric Nucleases Function For Ef Ficient Genome EditingFeng YueNo ratings yet
- Jove 138 57560Document8 pagesJove 138 57560lauyehsiangNo ratings yet
- N Comms 13274Document7 pagesN Comms 13274doniNo ratings yet
- 10 1016@j TRSL 2015 09 008Document17 pages10 1016@j TRSL 2015 09 008hiwmacrigeeeNo ratings yet
- Research Problem Assignment Week 6Document3 pagesResearch Problem Assignment Week 6Pearlyn LauNo ratings yet
- Crispr Cas 1Document10 pagesCrispr Cas 1ABHISHEK SWARNAKARNo ratings yet
- Gene Editing Prime Time 10.1056@NEJMcibr1914271Document4 pagesGene Editing Prime Time 10.1056@NEJMcibr1914271MCuk2606No ratings yet
- CRISPR - Nov27 - 2023 - TaggedDocument55 pagesCRISPR - Nov27 - 2023 - TaggedRitwik DasNo ratings yet
- 2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemDocument12 pages2021-Review-Single-Base Resolution Increasing The Specificity of The CRISPR-Cas SystemCristian Felipe Sandoval QuiñonezNo ratings yet
- 3 Gene Editing DocumentDocument4 pages3 Gene Editing DocumentAnanya ChauhanNo ratings yet
- CRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumDocument11 pagesCRISPR/Cas9 Mediated Targeting of Multiple Genes in DictyosteliumIram AmmadNo ratings yet
- Targeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesDocument21 pagesTargeted Genome Editing With A DNA-dependent DNA Polymerase and Exogenous DNA-containing TemplatesSabranth GuptaNo ratings yet
- Genome EditingDocument12 pagesGenome EditingDahlia StudioNo ratings yet
- NEB Featurearticle Crispr Cas9 Summer2014 LowresDocument6 pagesNEB Featurearticle Crispr Cas9 Summer2014 LowresRakhee RachelNo ratings yet
- A Protocol For Designing Sirnas With High Functionality and SpecificityDocument11 pagesA Protocol For Designing Sirnas With High Functionality and SpecificityThoha AlbaarNo ratings yet
- CRISPR Activation and Interference As Investigative ToolsDocument8 pagesCRISPR Activation and Interference As Investigative ToolsannyoryaNo ratings yet
- Genome Editing and Crispr Cas 9Document12 pagesGenome Editing and Crispr Cas 9HemsaiYadavNo ratings yet
- CRISPRDocument15 pagesCRISPRanirbanmanna88320No ratings yet
- Nobel Prize in Chemistry 2020: Institute of Agriculture and Animal Sciences (Iaas)Document5 pagesNobel Prize in Chemistry 2020: Institute of Agriculture and Animal Sciences (Iaas)Ramesh JØshiNo ratings yet
- A Review On The Mechanism and Applications of CRISPR Cas9 Cas12 Cas13 Cas14 Proteins Utilized For Genome EngineeringDocument15 pagesA Review On The Mechanism and Applications of CRISPR Cas9 Cas12 Cas13 Cas14 Proteins Utilized For Genome EngineeringrmbichiNo ratings yet
- Ijms 25 00823Document21 pagesIjms 25 00823Maria ClaraNo ratings yet
- DNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensFrom EverandDNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensNo ratings yet
- IDT - The CRISPR Basics HandbookDocument48 pagesIDT - The CRISPR Basics Handbookthauwui86100% (1)
- Application of Genetics and Biotechnology For Improving Medicinal PlantsDocument21 pagesApplication of Genetics and Biotechnology For Improving Medicinal PlantsAracely FernandaNo ratings yet
- Synthetic BiologyDocument38 pagesSynthetic BiologyHarry DouglasNo ratings yet
- ZFNDocument20 pagesZFNSenthilkumar PalanisamyNo ratings yet
- CRISPR 101 Ebook PDFDocument29 pagesCRISPR 101 Ebook PDFMarco Antonio Diaz GonzalezNo ratings yet
- Prahaar 3.0 Science SummaryDocument35 pagesPrahaar 3.0 Science Summaryjeshwanth2305No ratings yet
- GENE EDITING. Final PDFDocument13 pagesGENE EDITING. Final PDFS.A. DeeptiNo ratings yet
- Genetically Modified OrganismsDocument7 pagesGenetically Modified OrganismsPrajwal PatilNo ratings yet
- Restriction EnzymesDocument15 pagesRestriction EnzymesCharisma MeromiNo ratings yet
- Marine Microalgae For Production of Biofuels and Chemicals: SciencedirectDocument10 pagesMarine Microalgae For Production of Biofuels and Chemicals: SciencedirectShampa SenNo ratings yet
- Caft ReportDocument26 pagesCaft Reportrushi tahakikNo ratings yet
- s12929 018 0425 5 PDFDocument18 pagess12929 018 0425 5 PDFSURYONONo ratings yet
- Splat BookDocument170 pagesSplat Booksunny kuttanNo ratings yet
- ZFN, Talens, Crispr PDFDocument9 pagesZFN, Talens, Crispr PDFjezelle lividNo ratings yet
- UC Opp No. 2 CRISPR PatentDocument65 pagesUC Opp No. 2 CRISPR PatentjsherkowNo ratings yet
- Recent Advances in Animal Nutrition and Metabolism, Guoyao WuDocument344 pagesRecent Advances in Animal Nutrition and Metabolism, Guoyao WuPatricia TorresNo ratings yet
- Genome Engineeering Using Crispr Cas 9 System: June 2019Document8 pagesGenome Engineeering Using Crispr Cas 9 System: June 2019Mihaela CrafcenkoNo ratings yet
- Precision Medicine, CRISPR, and Genome Engineering: Stephen H. Tsang EditorDocument180 pagesPrecision Medicine, CRISPR, and Genome Engineering: Stephen H. Tsang EditorVinay Sagar LNo ratings yet
- Genetic EngineeringDocument26 pagesGenetic EngineeringMichaelAbdonDomingoFavoNo ratings yet
- Advanced Therapeutics - 2021 - ShahryariDocument42 pagesAdvanced Therapeutics - 2021 - ShahryariShubham AnandNo ratings yet
- Bio ProjectDocument11 pagesBio ProjectKshitij SharmaNo ratings yet
- Genome Editing An Ethical ReviewDocument136 pagesGenome Editing An Ethical ReviewIvica KelamNo ratings yet
- CRISPR+101+eBook v2021Document20 pagesCRISPR+101+eBook v2021Ruan GonçalvesNo ratings yet
- EDICIÓN GENÉTICA CRISPRcasDocument50 pagesEDICIÓN GENÉTICA CRISPRcasMAKARENA JIMENEZ VILLEGASNo ratings yet
- Liver Disease in Alpha-1 Antitrypsin Deficiency: Current Approaches and Future DirectionsDocument10 pagesLiver Disease in Alpha-1 Antitrypsin Deficiency: Current Approaches and Future DirectionsEianni Patrice De JoseNo ratings yet
- Synthego Ebook Crispr 101 PDFDocument29 pagesSynthego Ebook Crispr 101 PDFRie R100% (1)
- 2023 - Utilization of Genome Editing For Livestock Resilience in Changing EnvironmentDocument7 pages2023 - Utilization of Genome Editing For Livestock Resilience in Changing Environmentsoltani59No ratings yet
- ZFN, TALEN, and CRISPR-Cas-based Methods For Genome EngineeringDocument9 pagesZFN, TALEN, and CRISPR-Cas-based Methods For Genome EngineeringRomina Tamara Gil RamirezNo ratings yet
- Genetic Engineering of Microalgae For Enhanced Biorefi - 2020 - BiotechnologyDocument13 pagesGenetic Engineering of Microalgae For Enhanced Biorefi - 2020 - BiotechnologyerosNo ratings yet
- Editing Plant Genomes With Crispr/Cas9: SciencedirectDocument9 pagesEditing Plant Genomes With Crispr/Cas9: SciencedirectAli HamzaNo ratings yet