Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5825)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (903)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Dr. Sebi CookbookDocument110 pagesDr. Sebi CookbookAnonymous oTtlhP100% (18)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Ebook On Feed Formulation EbookDocument17 pagesEbook On Feed Formulation EbookDada RasheedNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Cat Substituent 588Document6 pagesCat Substituent 588beloshita_88No ratings yet
- 07 PKU Protein Content of AA PimentelDocument7 pages07 PKU Protein Content of AA Pimentelbeloshita_88No ratings yet
- Pku Consens SUA 2000Document17 pagesPku Consens SUA 2000beloshita_88No ratings yet
- Phenylalanine MG Umol CorespondentaDocument1 pagePhenylalanine MG Umol Corespondentabeloshita_88No ratings yet
- European Society For Phenylketonuria Calls For Minimum Standard of Care Across Europe - ENGLISHDocument3 pagesEuropean Society For Phenylketonuria Calls For Minimum Standard of Care Across Europe - ENGLISHbeloshita_88No ratings yet
- Response of Phenylketonuria To TetrahydrobiopterinDocument4 pagesResponse of Phenylketonuria To Tetrahydrobiopterinbeloshita_88No ratings yet
- Phenylalanine MG Umol CorespondentaDocument1 pagePhenylalanine MG Umol Corespondentabeloshita_88No ratings yet
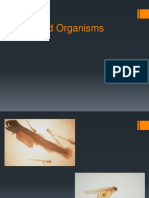
- Fish Food OrganismsDocument14 pagesFish Food OrganismsAmit SharmaNo ratings yet
- Unit 2 NutritionDocument28 pagesUnit 2 NutritionNescelle MacalintalNo ratings yet
- Metabolism Engineering Internship:: Health Bars For Disaster ReliefDocument19 pagesMetabolism Engineering Internship:: Health Bars For Disaster ReliefRick WuNo ratings yet
- Marquess I2B - Group 51Document14 pagesMarquess I2B - Group 51S BajpaiNo ratings yet
- Cacfp Menu AssessmentDocument2 pagesCacfp Menu Assessmentapi-604622509No ratings yet
- 2,000 Calorie Cutting Meal Plan (Male)Document2 pages2,000 Calorie Cutting Meal Plan (Male)usana.oneforallNo ratings yet
- Nutrition Final NoteDocument17 pagesNutrition Final Notediemtruc414No ratings yet
- Examples of Functional Child Outcomes-McWilliamDocument8 pagesExamples of Functional Child Outcomes-McWilliamKristin MurphyNo ratings yet
- Easy Vegan Pasta SaladDocument41 pagesEasy Vegan Pasta SaladDiana Madalina LuchianNo ratings yet
- Sem 1Document27 pagesSem 1hashikaNo ratings yet
- A Triumph of SurgeryDocument4 pagesA Triumph of SurgerySAHIL MANDAL 10i 12No ratings yet
- A Balanced Diet: Activity My Meals (Day 2)Document7 pagesA Balanced Diet: Activity My Meals (Day 2)Yaritza LVNo ratings yet
- NCP Excess Fluid VolumeDocument3 pagesNCP Excess Fluid VolumeJ.G RNo ratings yet
- Pea Protein Vs Whey ProteinDocument9 pagesPea Protein Vs Whey ProteinMiridarNo ratings yet
- Fitx Bonus - 8 Week Shred ProgramDocument10 pagesFitx Bonus - 8 Week Shred ProgramTHARNIKANTHNo ratings yet
- Calculation of Substrate Oxidation Rates in Vivo From Gaseous ExchangeDocument7 pagesCalculation of Substrate Oxidation Rates in Vivo From Gaseous ExchangeRodrigo CastilloNo ratings yet
- Pathfit 1 Module 12 Week 14Document2 pagesPathfit 1 Module 12 Week 14Mark Anthony RabyNo ratings yet
- Aristocrats Jaw WisdomDocument101 pagesAristocrats Jaw Wisdommr. manNo ratings yet
- Type 2 Diabetes Thesis PDFDocument5 pagesType 2 Diabetes Thesis PDFCollegePapersToBuyUK100% (2)
- Cosrx 66TH Prioritas SriDocument2 pagesCosrx 66TH Prioritas Srigandy.2237No ratings yet
- Plastipak ThermaLite Datasheet PDFDocument1 pagePlastipak ThermaLite Datasheet PDF武田慎也No ratings yet
- Bahasa InggrisDocument7 pagesBahasa InggrisdithobedaNo ratings yet
- TrueBasics RangeDocument71 pagesTrueBasics RangeLucky KhandelwalNo ratings yet
- G29 Data SheetDocument2 pagesG29 Data SheetDaniel TrujilloNo ratings yet
- Disgusting Food MuseumDocument2 pagesDisgusting Food Museum양현서No ratings yet
- State-Of-The-Art Process Technology For: MaizeDocument8 pagesState-Of-The-Art Process Technology For: MaizeMaria Camila ArenasNo ratings yet
- Figures-Facts-Benefits BENEO FinalDocument16 pagesFigures-Facts-Benefits BENEO FinalBryan TorresNo ratings yet
- Tabelul Periodic - Google SearchDocument1 pageTabelul Periodic - Google SearchMariaNo ratings yet