
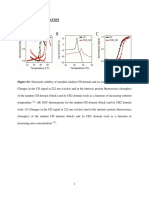
Acs Omega 2020
Acs Omega 2020
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5823)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Pre-Purchase Final Report Sea LightDocument46 pagesPre-Purchase Final Report Sea LightCESAR VIECNTE100% (1)
- Encyclopedia of Korean Folk BeliefsDocument167 pagesEncyclopedia of Korean Folk BeliefsAria7No ratings yet
- References ASEAN2Document3 pagesReferences ASEAN2M IrwaniNo ratings yet
- Bi5b00969 Si 001Document4 pagesBi5b00969 Si 001Jon RumbleyNo ratings yet
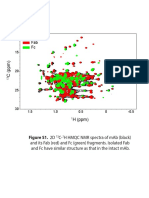
- Fab FC Mab: Figure S1. 2DDocument3 pagesFab FC Mab: Figure S1. 2DJon RumbleyNo ratings yet
- M-Value M-Value: 0 Unf M 0 Unf MDocument6 pagesM-Value M-Value: 0 Unf M 0 Unf MJon RumbleyNo ratings yet
- Surinder M. Singh, Swati Bandi, Dinen D. Shah, Geoffrey Armstrong, Krishna M. G. MallelaDocument9 pagesSurinder M. Singh, Swati Bandi, Dinen D. Shah, Geoffrey Armstrong, Krishna M. G. MallelaJon RumbleyNo ratings yet
- Kaong PaperDocument10 pagesKaong PapermikxendyNo ratings yet
- Narrative Text - Infografis - KurmerDocument2 pagesNarrative Text - Infografis - KurmerCitra Arba RojunaNo ratings yet
- Construction Method (PICC)Document2 pagesConstruction Method (PICC)rheymar diwaNo ratings yet
- Buttermilk Pancakes and Honey Roasted Figs Recipe HelloFreshDocument1 pageButtermilk Pancakes and Honey Roasted Figs Recipe HelloFreshxarogi1776No ratings yet
- Iui Made Easy: Semen Analysis, Processing and PreservationDocument97 pagesIui Made Easy: Semen Analysis, Processing and PreservationSuryakant HayatnagarkarNo ratings yet
- Antares Eng Rev02Document2 pagesAntares Eng Rev02Steven BrownNo ratings yet
- Fortran CF DDocument160 pagesFortran CF DLahcen AkerkouchNo ratings yet
- Experiment No 02 (A)Document3 pagesExperiment No 02 (A)Md Sabbir HossainNo ratings yet
- Starch BasedDocument6 pagesStarch BasedBongzkie Irudistan AmanteNo ratings yet
- PPDocument15 pagesPPdana angela hernandezNo ratings yet
- PPAP Workbook SupplierDocument25 pagesPPAP Workbook SupplierJuan VillaNo ratings yet
- Grammar Exam Pre Int PDFDocument2 pagesGrammar Exam Pre Int PDFSandra Lazovska100% (1)
- The Scientific MethodDocument2 pagesThe Scientific MethodArnold Alos100% (2)
- BEEIE - Unit 4 & 5 Question BankDocument7 pagesBEEIE - Unit 4 & 5 Question Banksachin barathNo ratings yet
- 20 Types of Pasta - TLE9Document2 pages20 Types of Pasta - TLE9Chloe RaniaNo ratings yet
- Asahi Carbon Black (For Rubber) Physical Chemistry Properties of Main Products - Products - Products and Technology - ASAHI CARBON CO., LTDDocument1 pageAsahi Carbon Black (For Rubber) Physical Chemistry Properties of Main Products - Products - Products and Technology - ASAHI CARBON CO., LTDSUDARSHAN dAWNo ratings yet
- FarmMachineryEquipment I ManualDocument51 pagesFarmMachineryEquipment I ManualBea SmithNo ratings yet
- Banco Preguntas Prueba SaberDocument21 pagesBanco Preguntas Prueba SaberYulieth Valeria Sanchez CortesNo ratings yet
- Effects of Maize-Legume Intercrops On Termite Damage To Maize, Activityof Predatoryants and Maize Yields in UgandaDocument7 pagesEffects of Maize-Legume Intercrops On Termite Damage To Maize, Activityof Predatoryants and Maize Yields in UgandasedianpoNo ratings yet
- Zeiss Erosion ModuleDocument13 pagesZeiss Erosion ModulepakhiddeyasNo ratings yet
- So-Called Logical Levels WoodsmallDocument29 pagesSo-Called Logical Levels WoodsmallDan Paris100% (2)
- Differential Equations MSC MathematicsDocument2 pagesDifferential Equations MSC MathematicsSamadNo ratings yet
- Brown Et Al. 2009 Tribolium A Model For Developmental and Pest BiologyDocument9 pagesBrown Et Al. 2009 Tribolium A Model For Developmental and Pest BiologyAneel Nizar AliNo ratings yet
- Renoise User ManualDocument198 pagesRenoise User Manualdrkstr77No ratings yet
- 03051Document2 pages03051JojolasNo ratings yet
- DLL Science 9 JulyDocument10 pagesDLL Science 9 JulyMark Kiven MartinezNo ratings yet
- PRC General Edu A 2007Document48 pagesPRC General Edu A 2007Dhena H Rasul SabdulaNo ratings yet









































































Download as pdf or txt
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5823)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1093)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (852)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (898)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (541)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (349)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (823)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (403)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Pre-Purchase Final Report Sea LightDocument46 pagesPre-Purchase Final Report Sea LightCESAR VIECNTE100% (1)
- Encyclopedia of Korean Folk BeliefsDocument167 pagesEncyclopedia of Korean Folk BeliefsAria7No ratings yet
- References ASEAN2Document3 pagesReferences ASEAN2M IrwaniNo ratings yet
- Bi5b00969 Si 001Document4 pagesBi5b00969 Si 001Jon RumbleyNo ratings yet
- Fab FC Mab: Figure S1. 2DDocument3 pagesFab FC Mab: Figure S1. 2DJon RumbleyNo ratings yet
- M-Value M-Value: 0 Unf M 0 Unf MDocument6 pagesM-Value M-Value: 0 Unf M 0 Unf MJon RumbleyNo ratings yet
- Surinder M. Singh, Swati Bandi, Dinen D. Shah, Geoffrey Armstrong, Krishna M. G. MallelaDocument9 pagesSurinder M. Singh, Swati Bandi, Dinen D. Shah, Geoffrey Armstrong, Krishna M. G. MallelaJon RumbleyNo ratings yet
- Kaong PaperDocument10 pagesKaong PapermikxendyNo ratings yet
- Narrative Text - Infografis - KurmerDocument2 pagesNarrative Text - Infografis - KurmerCitra Arba RojunaNo ratings yet
- Construction Method (PICC)Document2 pagesConstruction Method (PICC)rheymar diwaNo ratings yet
- Buttermilk Pancakes and Honey Roasted Figs Recipe HelloFreshDocument1 pageButtermilk Pancakes and Honey Roasted Figs Recipe HelloFreshxarogi1776No ratings yet
- Iui Made Easy: Semen Analysis, Processing and PreservationDocument97 pagesIui Made Easy: Semen Analysis, Processing and PreservationSuryakant HayatnagarkarNo ratings yet
- Antares Eng Rev02Document2 pagesAntares Eng Rev02Steven BrownNo ratings yet
- Fortran CF DDocument160 pagesFortran CF DLahcen AkerkouchNo ratings yet
- Experiment No 02 (A)Document3 pagesExperiment No 02 (A)Md Sabbir HossainNo ratings yet
- Starch BasedDocument6 pagesStarch BasedBongzkie Irudistan AmanteNo ratings yet
- PPDocument15 pagesPPdana angela hernandezNo ratings yet
- PPAP Workbook SupplierDocument25 pagesPPAP Workbook SupplierJuan VillaNo ratings yet
- Grammar Exam Pre Int PDFDocument2 pagesGrammar Exam Pre Int PDFSandra Lazovska100% (1)
- The Scientific MethodDocument2 pagesThe Scientific MethodArnold Alos100% (2)
- BEEIE - Unit 4 & 5 Question BankDocument7 pagesBEEIE - Unit 4 & 5 Question Banksachin barathNo ratings yet
- 20 Types of Pasta - TLE9Document2 pages20 Types of Pasta - TLE9Chloe RaniaNo ratings yet
- Asahi Carbon Black (For Rubber) Physical Chemistry Properties of Main Products - Products - Products and Technology - ASAHI CARBON CO., LTDDocument1 pageAsahi Carbon Black (For Rubber) Physical Chemistry Properties of Main Products - Products - Products and Technology - ASAHI CARBON CO., LTDSUDARSHAN dAWNo ratings yet
- FarmMachineryEquipment I ManualDocument51 pagesFarmMachineryEquipment I ManualBea SmithNo ratings yet
- Banco Preguntas Prueba SaberDocument21 pagesBanco Preguntas Prueba SaberYulieth Valeria Sanchez CortesNo ratings yet
- Effects of Maize-Legume Intercrops On Termite Damage To Maize, Activityof Predatoryants and Maize Yields in UgandaDocument7 pagesEffects of Maize-Legume Intercrops On Termite Damage To Maize, Activityof Predatoryants and Maize Yields in UgandasedianpoNo ratings yet
- Zeiss Erosion ModuleDocument13 pagesZeiss Erosion ModulepakhiddeyasNo ratings yet
- So-Called Logical Levels WoodsmallDocument29 pagesSo-Called Logical Levels WoodsmallDan Paris100% (2)
- Differential Equations MSC MathematicsDocument2 pagesDifferential Equations MSC MathematicsSamadNo ratings yet
- Brown Et Al. 2009 Tribolium A Model For Developmental and Pest BiologyDocument9 pagesBrown Et Al. 2009 Tribolium A Model For Developmental and Pest BiologyAneel Nizar AliNo ratings yet
- Renoise User ManualDocument198 pagesRenoise User Manualdrkstr77No ratings yet
- 03051Document2 pages03051JojolasNo ratings yet
- DLL Science 9 JulyDocument10 pagesDLL Science 9 JulyMark Kiven MartinezNo ratings yet
- PRC General Edu A 2007Document48 pagesPRC General Edu A 2007Dhena H Rasul SabdulaNo ratings yet