Download as pdf or txt
You might also like
- Republican Challenge Against Mail Ballot LawDocument18 pagesRepublican Challenge Against Mail Ballot LawJessica HillNo ratings yet
- Afimilk MPC User ManualDocument43 pagesAfimilk MPC User ManuallegarciNo ratings yet
- Red Wizard's Dream 2 - Dead in Thay PDFDocument107 pagesRed Wizard's Dream 2 - Dead in Thay PDFtetris479100% (4)
- Business Plan of MushroomsDocument29 pagesBusiness Plan of MushroomsMahamudul Hasan80% (5)
- Choanal Atresia1Document5 pagesChoanal Atresia1anurajoneNo ratings yet
- Nihms 1657970Document20 pagesNihms 1657970mchojnacki81No ratings yet
- CNLDO JurnalDocument6 pagesCNLDO JurnalKhairul FitrahNo ratings yet
- ENT Pediatri AnesDocument5 pagesENT Pediatri AnesStanley SuhermanNo ratings yet
- Anaesthesia For Paediatric Ear, Nose, and Throat Surgery 2007Document5 pagesAnaesthesia For Paediatric Ear, Nose, and Throat Surgery 2007zaftotNo ratings yet
- Upper Airway Obstruction in Children: Review ArticleDocument8 pagesUpper Airway Obstruction in Children: Review ArticleLis RibeiroNo ratings yet
- Choanal AtresiaDocument8 pagesChoanal AtresiaDantowaluyo NewNo ratings yet
- Ear, Nose and Throat EmergenciesDocument3 pagesEar, Nose and Throat Emergenciesfmta100% (1)
- Choanal Atresia PDFDocument8 pagesChoanal Atresia PDFMonna Medani LysabellaNo ratings yet
- Seminar On Choanal AtresiaDocument16 pagesSeminar On Choanal AtresiaGreeshma ShajiNo ratings yet
- Choanal AtresiaDocument32 pagesChoanal AtresiaGreeshma ShajiNo ratings yet
- Choanal AtresiaDocument5 pagesChoanal AtresiaPriyaNo ratings yet
- Uia30 Anaesthesia For Paediatric Ear Nose and Throat SurgeryDocument6 pagesUia30 Anaesthesia For Paediatric Ear Nose and Throat SurgeryAshokNo ratings yet
- 2022 Nasal Pyriform Aperture StenosisDocument2 pages2022 Nasal Pyriform Aperture StenosisNicolien van der PoelNo ratings yet
- Preauricular Sinus and Its ManagmentDocument12 pagesPreauricular Sinus and Its ManagmentDr. T. Balasubramanian100% (7)
- Choanal Atresia, Epistaxis & AspirationDocument12 pagesChoanal Atresia, Epistaxis & Aspirationsubinj_350% (2)
- Adenoid Hypertrophy Adenoidectomy Cesar GarciaDocument56 pagesAdenoid Hypertrophy Adenoidectomy Cesar GarciaIemima RotunduNo ratings yet
- File 18584Document12 pagesFile 18584Mohammed MuthanaNo ratings yet
- Ent Throat 1Document58 pagesEnt Throat 1ميمونه عبدالرحيم مصطفىNo ratings yet
- Adenoid Hypertrophy FixDocument7 pagesAdenoid Hypertrophy FixWidi Yuli HariantoNo ratings yet
- Airway Obstruction Final2Document33 pagesAirway Obstruction Final2Mahindra KumarNo ratings yet
- Choanal AtresiaDocument3 pagesChoanal AtresiaKunal GowdaNo ratings yet
- Managing NosebleedsDocument7 pagesManaging NosebleedsHasbiallah YusufNo ratings yet
- Respiratory SystemDocument24 pagesRespiratory SystemHani El-asferNo ratings yet
- Airway AnomaliesDocument11 pagesAirway AnomaliesDariki CampbellNo ratings yet
- VacioDocument17 pagesVacioKaren HolguinNo ratings yet
- Coanal AtresiaDocument4 pagesCoanal AtresiaBkas GrgNo ratings yet
- Anatomy of Nasopharynx & AdenoidsDocument3 pagesAnatomy of Nasopharynx & AdenoidsAmy KochNo ratings yet
- Cleft PalateDocument16 pagesCleft PalateAnu Priya GopuNo ratings yet
- Pediatric Airway Disorders and Parenchymal Lung DiseaseDocument10 pagesPediatric Airway Disorders and Parenchymal Lung DiseasePuma ChanNo ratings yet
- Kelainan Kongenital Sistem RespirasiDocument54 pagesKelainan Kongenital Sistem RespirasiBhima PerdanaNo ratings yet
- Presentation On Cogenital Anomalies of Respiratory TractDocument89 pagesPresentation On Cogenital Anomalies of Respiratory Tractmaggykariuki002No ratings yet
- Pediatric Laryngotracheal Stenosis and Airway Reconstruction: A Review of Voice Outcomes, Assessment, and Treatment IssuesDocument11 pagesPediatric Laryngotracheal Stenosis and Airway Reconstruction: A Review of Voice Outcomes, Assessment, and Treatment Issuescanndy202No ratings yet
- Throat Pharynx Adenoids ENT LecturesDocument33 pagesThroat Pharynx Adenoids ENT LecturesRaju GangadharanNo ratings yet
- Otitis Media With EffusionDocument3 pagesOtitis Media With EffusionAnish RajNo ratings yet
- Cleft Lip and PalateDocument10 pagesCleft Lip and Palatehiwotbetesfa2123No ratings yet
- Article in Press: Nasal Obstruction in ChildrenDocument4 pagesArticle in Press: Nasal Obstruction in ChildrenLimeysahni NazhoeNo ratings yet
- Congenital AnomaliesDocument22 pagesCongenital Anomaliesjessy100% (1)
- Chapter 5 - Adenoidectomy: Suman GollaDocument7 pagesChapter 5 - Adenoidectomy: Suman GollaGloriaCorredorNo ratings yet
- Laryngomalacia PDFDocument6 pagesLaryngomalacia PDFFilologus SiwabessyNo ratings yet
- Chapter 027Document3 pagesChapter 027barbaraNo ratings yet
- Cap. 12. ENT DISORDERS PDFDocument10 pagesCap. 12. ENT DISORDERS PDFoana policarpov100% (1)
- Atlas of Paranasal Sinus SurgeryDocument81 pagesAtlas of Paranasal Sinus SurgeryNemer Al-KhtoumNo ratings yet
- Choanal Atresia SDocument4 pagesChoanal Atresia SCosbyNo ratings yet
- Laryngomalaci 2016 Ajoshua Bedwell, MD, GeorgeZalzal, MDNDocument4 pagesLaryngomalaci 2016 Ajoshua Bedwell, MD, GeorgeZalzal, MDNwawa chenNo ratings yet
- Tonsilectomy and AdenoidectomyDocument7 pagesTonsilectomy and AdenoidectomySabella Gustika VernandaNo ratings yet
- Atrezia Coanala Este o Conditie Congenitala in Care Regiunea Posterioara A Pasajului Nazal (Coana) Este Blocata, de ObiceiDocument5 pagesAtrezia Coanala Este o Conditie Congenitala in Care Regiunea Posterioara A Pasajului Nazal (Coana) Este Blocata, de ObiceiTatiana NegrituNo ratings yet
- Ear Nose and Throat EmergenciesDocument3 pagesEar Nose and Throat EmergenciesHeribertus AribawaNo ratings yet
- ManagingAirwayObstruction PDFDocument5 pagesManagingAirwayObstruction PDFMeyNo ratings yet
- Choanal Atresia 2014 01 BDocument10 pagesChoanal Atresia 2014 01 BandisNo ratings yet
- Cleft Lip Andpalate SurgeryDocument4 pagesCleft Lip Andpalate Surgerydr.rrajesh92No ratings yet
- 02 Acute Airway ObstructionDocument3 pages02 Acute Airway Obstructioncharmainemargaret.parreno.medNo ratings yet
- Nasal Polyp MedscapeDocument30 pagesNasal Polyp MedscapeMonissa ArrianiNo ratings yet
- Fernandez D. Snoring and Obstructive Apnea, Upper Airway. Emedicine Last Update April, 27 TH 2015. Available From: URLDocument12 pagesFernandez D. Snoring and Obstructive Apnea, Upper Airway. Emedicine Last Update April, 27 TH 2015. Available From: URLHari SubagiyoNo ratings yet
- Estenosis Subglotica 2Document18 pagesEstenosis Subglotica 2Nicolás HenaoNo ratings yet
- Congenital Bilateral Choanal AtresiDocument5 pagesCongenital Bilateral Choanal AtresiMonna Medani LysabellaNo ratings yet
- Desarrollo Via AereaDocument5 pagesDesarrollo Via AereaLudy Jiménez ValdiviaNo ratings yet
- Krespi 1997Document4 pagesKrespi 1997kwpang1No ratings yet
- Special Situations in Management of Tonsil and Adenoid DisordersDocument49 pagesSpecial Situations in Management of Tonsil and Adenoid DisordersEugene AlexNo ratings yet
- Bone-Age-Bookgreylis PyleDocument61 pagesBone-Age-Bookgreylis PyleadeNo ratings yet
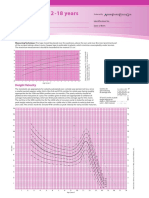
- Girls-2-18 YearsDocument4 pagesGirls-2-18 YearsadeNo ratings yet
- Boys-2-18 YearsDocument4 pagesBoys-2-18 YearsadeNo ratings yet
- SOP 6-Minute-Walk Test 1. General Considerations: 6-Minute Walking Distance: Covered Distance inDocument5 pagesSOP 6-Minute-Walk Test 1. General Considerations: 6-Minute Walking Distance: Covered Distance inadeNo ratings yet
- Percutaneous Catheter Evacuation of A Pneumatocele in An Extremely Premature Infant With Respiratory FailureDocument3 pagesPercutaneous Catheter Evacuation of A Pneumatocele in An Extremely Premature Infant With Respiratory FailureadeNo ratings yet
- Fitzsimons 2014 Allergic DiseaseDocument11 pagesFitzsimons 2014 Allergic DiseaseadeNo ratings yet
- Evaluation of Functional Capacity For Exercise in Children and Adolescents With Sickle-Cell Disease Through The Six-Minute Walk TestDocument7 pagesEvaluation of Functional Capacity For Exercise in Children and Adolescents With Sickle-Cell Disease Through The Six-Minute Walk TestadeNo ratings yet
- Pone 0108922Document5 pagesPone 0108922adeNo ratings yet
- A, Et Al. M I N S: Web Table I PDocument1 pageA, Et Al. M I N S: Web Table I PadeNo ratings yet
- Pone 0108922Document5 pagesPone 0108922adeNo ratings yet
- 81 MantapDocument8 pages81 MantapadeNo ratings yet
- 10.1177@0009922820908583SBI Faktor InfeksiDocument7 pages10.1177@0009922820908583SBI Faktor InfeksiadeNo ratings yet
- Pediatric Steroid Dependence Nephrotic Syndrome: A Case SeriesDocument2 pagesPediatric Steroid Dependence Nephrotic Syndrome: A Case SeriesadeNo ratings yet
- Habib y 1995Document4 pagesHabib y 1995adeNo ratings yet
- Neonates in The COVID-19 Pandemic: EditorialDocument3 pagesNeonates in The COVID-19 Pandemic: EditorialadeNo ratings yet
- Letter To The Editor: Neonatal Coronavirus Disease-2019: Presentation and ManagementDocument3 pagesLetter To The Editor: Neonatal Coronavirus Disease-2019: Presentation and ManagementadeNo ratings yet
- Neonatal Covid 19 Exposures and Infections A Systematic Review of Modes of Transmission Manifestations and ManagementDocument18 pagesNeonatal Covid 19 Exposures and Infections A Systematic Review of Modes of Transmission Manifestations and ManagementadeNo ratings yet
- WHO Update On Case Management - Regina LoprangDocument25 pagesWHO Update On Case Management - Regina LoprangadeNo ratings yet
- Critical Care Beyond The Picu, Since The Emergency Room: How Come The Pediatric Intensivist Be A Covidologist?Document44 pagesCritical Care Beyond The Picu, Since The Emergency Room: How Come The Pediatric Intensivist Be A Covidologist?adeNo ratings yet
- Guidelines For Neonates of Mothers - 1301-NewDocument5 pagesGuidelines For Neonates of Mothers - 1301-NewadeNo ratings yet
- Weiser 2011Document3 pagesWeiser 2011adeNo ratings yet
- Investigation of Small Dams Failures in Seasonal/Intermittent Streams: Unpublished PapersDocument33 pagesInvestigation of Small Dams Failures in Seasonal/Intermittent Streams: Unpublished Papersוויסאם חטארNo ratings yet
- YOUTH AbbreviationDocument23 pagesYOUTH AbbreviationKen ElectronNo ratings yet
- Biogas From Livestock WasteDocument19 pagesBiogas From Livestock WasteRosmawatiNo ratings yet
- BPPO Complan Report March 25, 2016Document10 pagesBPPO Complan Report March 25, 2016Benguet PpoNo ratings yet
- Bec ExaminationDocument2 pagesBec ExaminationHiken D. ChoNo ratings yet
- Percakapan Bahasa Inggris SMP Negeri 1 MaduranDocument2 pagesPercakapan Bahasa Inggris SMP Negeri 1 MaduranDevi WulansariNo ratings yet
- HMP Openser Sip AnDocument9 pagesHMP Openser Sip Anchingo9252No ratings yet
- Major in Social Studies Room AssignmentDocument24 pagesMajor in Social Studies Room AssignmentPRC Baguio100% (1)
- Literature Review On Attitude Towards MathematicsDocument10 pagesLiterature Review On Attitude Towards MathematicsafdtveepoNo ratings yet
- Export Act 1963Document27 pagesExport Act 1963Anonymous OPix6Tyk5INo ratings yet
- Media BiasDocument66 pagesMedia BiasAIG at ECU0% (1)
- Distored SchemasDocument14 pagesDistored SchemasJoelNo ratings yet
- Spouting Lore - Playing Stonetop (And Other PbtA Games)Document30 pagesSpouting Lore - Playing Stonetop (And Other PbtA Games)Gabriel KerrNo ratings yet
- Survey Letter July 17Document2 pagesSurvey Letter July 17Michael ScarfoNo ratings yet
- Days of Wine and Roses - For SymphonyDocument1 pageDays of Wine and Roses - For SymphonyEmmett Amitai WolbergerNo ratings yet
- SBP - Pengenalan, Induksi RulesDocument37 pagesSBP - Pengenalan, Induksi RulesTU ElektroNo ratings yet
- Mecha v. INS, 4th Cir. (2000)Document4 pagesMecha v. INS, 4th Cir. (2000)Scribd Government DocsNo ratings yet
- Pedodontic Lect 20-21Document8 pagesPedodontic Lect 20-21كوثر علي عدنان حسينNo ratings yet
- US Open 2022 QuizDocument2 pagesUS Open 2022 QuizNelson D'SouzaNo ratings yet
- Service Marketing: BY, Bhavithra AsokanDocument20 pagesService Marketing: BY, Bhavithra AsokanDomnic SavioNo ratings yet
- Solid Triangle Sales Corp. v. Sheriff of RTC QC, G. R. No. 144309, November 23,2001Document14 pagesSolid Triangle Sales Corp. v. Sheriff of RTC QC, G. R. No. 144309, November 23,2001Cza PeñaNo ratings yet
- COTDocument19 pagesCOTAF Dowell Mirin0% (1)
- Fungal NutritionDocument3 pagesFungal NutritionArtemishaMtzNo ratings yet
- Mini Paper Company Profile by Decky M.A (19110115)Document14 pagesMini Paper Company Profile by Decky M.A (19110115)Adelia FebriantiNo ratings yet
- Thirumurai 2Document188 pagesThirumurai 2thegodkannanNo ratings yet
- CAS QuizDocument6 pagesCAS QuizLeng ChhunNo ratings yet