Download as pdf or txt
You might also like
- Worksheet Activity No. 4: Preparation of Nutrient BrothDocument3 pagesWorksheet Activity No. 4: Preparation of Nutrient BrothPandangan MatiynNo ratings yet
- Biodegradation of Bioplastics in Natural EnvironmentsDocument11 pagesBiodegradation of Bioplastics in Natural EnvironmentsKaren Puente CedilloNo ratings yet
- Systematic Procedure For Qualitative Analysis of Inorganic Salts and MixturesDocument3 pagesSystematic Procedure For Qualitative Analysis of Inorganic Salts and MixturesSahithi Reddy K33% (3)
- Culture MediaDocument8 pagesCulture MediaHershey BaconNo ratings yet
- Chapter X Culture Media Preparation, InoculationDocument71 pagesChapter X Culture Media Preparation, InoculationBenyam ZenebeNo ratings yet
- Culture Media Preparation, InoculationDocument67 pagesCulture Media Preparation, InoculationGebrekidanhaftayNo ratings yet
- 10 Culture Media Preparation, Inoculation (2) (Autosaved)Document68 pages10 Culture Media Preparation, Inoculation (2) (Autosaved)Firaol ManNo ratings yet
- Lab 3Document17 pagesLab 3mahirrasho75No ratings yet
- Culture MediaDocument34 pagesCulture MediaIsak Isak IsakNo ratings yet
- Lab 2 Culture MediaDocument15 pagesLab 2 Culture Mediaتجربة أولىNo ratings yet
- Industrial Microbiology IDocument20 pagesIndustrial Microbiology ISharif JamsNo ratings yet
- Mku Bacterial GrowthDocument9 pagesMku Bacterial GrowthMandillah S EddieNo ratings yet
- 21030Document9 pages21030ARG ShovonNo ratings yet
- Bacteriological Culture Media: Practical No.: DateDocument3 pagesBacteriological Culture Media: Practical No.: Datekushal NeupaneNo ratings yet
- Culture Media and Cultivation of BacteriaDocument14 pagesCulture Media and Cultivation of BacteriaatharvatanksaleNo ratings yet
- INFORME 3 - MICROBIOLOGÍA - Mendoza MelanieDocument8 pagesINFORME 3 - MICROBIOLOGÍA - Mendoza MelanieMelanie Lucero Mendoza ChaparroNo ratings yet
- Culture Media - Classification, Types, and Relevance - Conduct ScienceDocument7 pagesCulture Media - Classification, Types, and Relevance - Conduct ScienceHaseeba KhanNo ratings yet
- Microbial Culture Media Definition: The Media Is A Source of Nutrients To Support The Growth of The Micro-Organisms inDocument10 pagesMicrobial Culture Media Definition: The Media Is A Source of Nutrients To Support The Growth of The Micro-Organisms inARG ShovonNo ratings yet
- 5 - CULTURE MEDIA AnimeshDocument35 pages5 - CULTURE MEDIA Animeshsouvikmaity2024No ratings yet
- Microbiology Assignment: Name: Laiba Ali Registartion No.: L1F20Bsft0040Document8 pagesMicrobiology Assignment: Name: Laiba Ali Registartion No.: L1F20Bsft0040Laiba AliNo ratings yet
- Activity 4culturemedia - CajesNDocument2 pagesActivity 4culturemedia - CajesNCAJES NOLINo ratings yet
- Chapter X Culture Media Preparation, Inoculation and IdentifDocument66 pagesChapter X Culture Media Preparation, Inoculation and IdentifMendy SolomonNo ratings yet
- MicrobioLab Lab 2 BS Bio 2CDocument3 pagesMicrobioLab Lab 2 BS Bio 2CLeslie Ann PotencianoNo ratings yet
- Culture MediaDocument11 pagesCulture MediaAbDul RehManNo ratings yet
- Serial No. Name of The Content Page NoDocument11 pagesSerial No. Name of The Content Page NoMd. Mohib UllahNo ratings yet
- Culture Media Used in Microbiology: Salman Tausif Senior Technologist Clinical MicrobiologyDocument36 pagesCulture Media Used in Microbiology: Salman Tausif Senior Technologist Clinical MicrobiologyZeeshan YousufNo ratings yet
- Media Preparation and Uses in Medical MicrobiologyDocument20 pagesMedia Preparation and Uses in Medical MicrobiologyPrincewill SeiyefaNo ratings yet
- Culture MediaDocument10 pagesCulture Mediaayeshasaleem107112No ratings yet
- 1491 1599547580 Fair Lab Manual - Micro S3Document32 pages1491 1599547580 Fair Lab Manual - Micro S3jerinbiju1019No ratings yet
- Lab 3 Media W23Document6 pagesLab 3 Media W23devaanshNo ratings yet
- Culture Media PreparationDocument29 pagesCulture Media PreparationKatrina Mae MedinaNo ratings yet
- Culture Media Presentation by DR Irfan Final VersionDocument24 pagesCulture Media Presentation by DR Irfan Final Versionyaseenahmad544No ratings yet
- Microbiology Lab ManualDocument52 pagesMicrobiology Lab ManualHà Anh Minh Lê100% (1)
- Culture MethodsDocument64 pagesCulture MethodsMisbah ShabbirNo ratings yet
- Microlab Indiv Act 5 Not ParaphrasedDocument12 pagesMicrolab Indiv Act 5 Not Paraphrasedmaricarcardenas04No ratings yet
- Laboratory Exercise 3. Culture Media Preparation and SterilizationDocument8 pagesLaboratory Exercise 3. Culture Media Preparation and SterilizationNesly Joy CaballeganNo ratings yet
- Culture MediaDocument5 pagesCulture MediaMuhammad Abu HurairaNo ratings yet
- Basic Culture MediaDocument7 pagesBasic Culture MediaramNo ratings yet
- Bacteriology (Methods of Studying Bacteria) : Anne Lorraine Magarette Dulay MLS 3.1 JULY 3, 2018Document15 pagesBacteriology (Methods of Studying Bacteria) : Anne Lorraine Magarette Dulay MLS 3.1 JULY 3, 2018Anne Lorraine Margarette DulayNo ratings yet
- CABERIO AB21 LABORATORY REPORT NO. 3 Microbio LABDocument5 pagesCABERIO AB21 LABORATORY REPORT NO. 3 Microbio LABJohn Mark Gallano CanayonNo ratings yet
- Reviewer in Microbiology and ParasitologyDocument13 pagesReviewer in Microbiology and ParasitologyCharlot Navarro100% (2)
- Microbiology NotesDocument9 pagesMicrobiology Notesshreevidya4gurunagesNo ratings yet
- Lab Manual Microbiology - 2023Document58 pagesLab Manual Microbiology - 2023do hieuNo ratings yet
- Experiment Preparation of Media: 7.0 ObjectivesDocument11 pagesExperiment Preparation of Media: 7.0 ObjectivesAnkush BiswasNo ratings yet
- PHBIOSCI 5B - ACTIVITY #1 - Basic Laboratory Techniques For Isolation, Cultivation, and Cultural Characterization of Microorganisms (SUMMER 2022)Document1 pagePHBIOSCI 5B - ACTIVITY #1 - Basic Laboratory Techniques For Isolation, Cultivation, and Cultural Characterization of Microorganisms (SUMMER 2022)Shopifyy ClothingNo ratings yet
- Lab Report 3 and 4Document22 pagesLab Report 3 and 4siyabongaminenhlemasekoNo ratings yet
- Bacterial Culture MediaDocument10 pagesBacterial Culture Medianosila_oz854No ratings yet
- Culture MediaDocument36 pagesCulture MediaPatricia Anne Nicole CuaresmaNo ratings yet
- Culture MediaDocument8 pagesCulture MediaAb AbNo ratings yet
- Microbiology Practical Copy 2022Document24 pagesMicrobiology Practical Copy 2022Saad AbidNo ratings yet
- M1 E6 Pre LabDocument2 pagesM1 E6 Pre LabApplePi SimpNo ratings yet
- Culturing of Bacteria and Culture MethodsDocument47 pagesCulturing of Bacteria and Culture MethodsQawiyy 55No ratings yet
- Bab I Pendahuluan 1.1. Latar BelakangDocument13 pagesBab I Pendahuluan 1.1. Latar BelakangRinalNo ratings yet
- Culture MediaDocument3 pagesCulture MediaReyhan EgeNo ratings yet
- Etu FM 222Document32 pagesEtu FM 222Bebi WakaNo ratings yet
- CULTURINGDocument12 pagesCULTURINGvictor mangataNo ratings yet
- Introduction To Lab 6: Ex. Preparation of Culture MediaDocument13 pagesIntroduction To Lab 6: Ex. Preparation of Culture Mediaশর্ট সার্কিটNo ratings yet
- CULTURINGDocument10 pagesCULTURINGpeterNo ratings yet
- Microbiology ReportDocument10 pagesMicrobiology ReportHassan MohammedNo ratings yet
- Practical 1 MediaDocument35 pagesPractical 1 MediaPatrisha BuanNo ratings yet
- LABORATORY MANUAL FOR A MINI PROJECT: MSCB 1113 BIOCHEMISTRY & MICROBIAL PHYSIOLOGYFrom EverandLABORATORY MANUAL FOR A MINI PROJECT: MSCB 1113 BIOCHEMISTRY & MICROBIAL PHYSIOLOGYNo ratings yet
- Food Selection and Preparation: A Laboratory ManualFrom EverandFood Selection and Preparation: A Laboratory ManualRating: 2.5 out of 5 stars2.5/5 (2)
- Internship Proposal-Đề cương Thực tậpDocument2 pagesInternship Proposal-Đề cương Thực tậpMinh DuyNo ratings yet
- Chap 1 Taking Risks and Making Profits Within The Dynamic Business EnvironmentDocument21 pagesChap 1 Taking Risks and Making Profits Within The Dynamic Business EnvironmentMinh DuyNo ratings yet
- Ref-1-Vietnam Food Law-2010-QH12Document37 pagesRef-1-Vietnam Food Law-2010-QH12Minh DuyNo ratings yet
- Lecture 3. Protein AnalysisDocument7 pagesLecture 3. Protein AnalysisMinh DuyNo ratings yet
- Cal2BT Final Spring2014 v2Document5 pagesCal2BT Final Spring2014 v2Minh DuyNo ratings yet
- Moisture and Total Solid Analysis: Content of This LectureDocument8 pagesMoisture and Total Solid Analysis: Content of This LectureMinh DuyNo ratings yet
- Pulling Together by John JDocument12 pagesPulling Together by John JMinh DuyNo ratings yet
- Lecture 2.2 Ash AnalysisDocument4 pagesLecture 2.2 Ash AnalysisMinh DuyNo ratings yet
- Food Analysis BTFT302IU Content of This Lecture: - Aim - AimDocument11 pagesFood Analysis BTFT302IU Content of This Lecture: - Aim - AimMinh DuyNo ratings yet
- Assignment of Lecture 2Document1 pageAssignment of Lecture 2Minh Duy100% (1)
- Chapter 16-Solid and Hazardous Waste: Multiple ChoiceDocument23 pagesChapter 16-Solid and Hazardous Waste: Multiple ChoiceMinh DuyNo ratings yet
- Food Laws and Food Standards: 1. General Course InformationDocument6 pagesFood Laws and Food Standards: 1. General Course InformationMinh DuyNo ratings yet
- Tien Duc: Unofficial SolutionDocument4 pagesTien Duc: Unofficial SolutionMinh DuyNo ratings yet
- W = F d = Fdcos (θ) : Unit: J Unit: J/s = WDocument14 pagesW = F d = Fdcos (θ) : Unit: J Unit: J/s = WMinh DuyNo ratings yet
- 3 1 47 813 PDFDocument4 pages3 1 47 813 PDFMinh DuyNo ratings yet
- A Comparative Pharmaceutico-Analytical Study of Punarnasava and PunarnarishtaDocument5 pagesA Comparative Pharmaceutico-Analytical Study of Punarnasava and Punarnarishtaalnrmamckoppa19No ratings yet
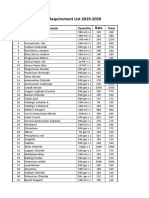
- Chemistry Requirement List 2019-2020: SR - No Chemicals Quantity TotalDocument2 pagesChemistry Requirement List 2019-2020: SR - No Chemicals Quantity TotalvaradNo ratings yet
- Systematic Qualitative Organic AnalysisDocument17 pagesSystematic Qualitative Organic Analysisravi@laviNo ratings yet
- Common Fatty AcidsDocument2 pagesCommon Fatty AcidsJohnNo ratings yet
- Class 6 Notes (Chapter 5)Document3 pagesClass 6 Notes (Chapter 5)Saman KhanNo ratings yet
- Chemical Plaque ControlDocument39 pagesChemical Plaque ControlSahin mollickNo ratings yet
- Structure of Atom AssignmentDocument20 pagesStructure of Atom Assignmentrajesh duaNo ratings yet
- Reactivity of Metal Complexes NotesDocument8 pagesReactivity of Metal Complexes Notesjyothi sai sriNo ratings yet
- Macromolecules: Self-Preparation Biology Assessment TestDocument36 pagesMacromolecules: Self-Preparation Biology Assessment Testmay ann dimaanoNo ratings yet
- Laboratoey & Chemical Analysis ProtienDocument98 pagesLaboratoey & Chemical Analysis Protienarifuzzaman ashadNo ratings yet
- Unit 4 Question and AnswersDocument14 pagesUnit 4 Question and AnswersSanjay KumarNo ratings yet
- 4 Colligative Properties of SolutionsDocument84 pages4 Colligative Properties of SolutionsujargohandiNo ratings yet
- SA FET Y#: Technical Data SheetDocument3 pagesSA FET Y#: Technical Data SheetDevi Gusni YantiNo ratings yet
- Thioflex 600 Gun Grade GreyDocument10 pagesThioflex 600 Gun Grade GreyAbbasNo ratings yet
- BlueInGreen SDOX CS Brochure v6Document2 pagesBlueInGreen SDOX CS Brochure v6miguel_vera6592No ratings yet
- Triethyl CitrateDocument14 pagesTriethyl CitrateEyad WaleedNo ratings yet
- Hydrodesulfurization: HistoryDocument5 pagesHydrodesulfurization: HistoryHevin HassanNo ratings yet
- The Notes On Histochemical StainsDocument125 pagesThe Notes On Histochemical StainsDr Joswin Dsa75% (4)
- PETE 311 Lab 1 MemoDocument3 pagesPETE 311 Lab 1 MemoTyler MroskoNo ratings yet
- Topic II Basic Principles of Extraction of Metals From Ores & PurificationDocument31 pagesTopic II Basic Principles of Extraction of Metals From Ores & PurificationKing of KingsNo ratings yet
- Olson Et Al. - 1981 - Crystal Structure and Structure-Related Properties of ZSM-5Document6 pagesOlson Et Al. - 1981 - Crystal Structure and Structure-Related Properties of ZSM-5Hari NarayananNo ratings yet
- Ncert Solutions Class 12 Chemistry Chapter 9 Coordination CompoundsDocument11 pagesNcert Solutions Class 12 Chemistry Chapter 9 Coordination Compoundspriya yadavNo ratings yet
- SN1 SN2 E1 E2 Reaction PHR-122Document36 pagesSN1 SN2 E1 E2 Reaction PHR-122zakariansu67% (6)
- Morphology and Classification of BacteriaDocument20 pagesMorphology and Classification of Bacteriavineetvishal73100% (1)
- Saponification: Esters, Soapless and Soapy DetergentsDocument17 pagesSaponification: Esters, Soapless and Soapy Detergentsp bergerNo ratings yet
- Common Name Vs Chemical NameDocument5 pagesCommon Name Vs Chemical NameManeesh RanjanNo ratings yet
- Construction and Building Materials: Salvador Ordóñez, Ángel La Iglesia, Miguel Louis, M Ángeles García-del-CuraDocument12 pagesConstruction and Building Materials: Salvador Ordóñez, Ángel La Iglesia, Miguel Louis, M Ángeles García-del-CuraLiLiJin WookNo ratings yet
- Chapter 3 - Igneous Rocks PDFDocument31 pagesChapter 3 - Igneous Rocks PDFmahmoud alawnehNo ratings yet