
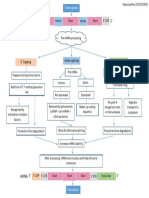
Eukaryotic Genome Complexity - Learn Science at Scitable
Eukaryotic Genome Complexity - Learn Science at Scitable
You might also like
- Merritt's BakeryDocument1 pageMerritt's BakeryNardine Farag0% (1)
- Cambridge IGCSE Chemistry Coursebook With CD-ROM PDFDocument532 pagesCambridge IGCSE Chemistry Coursebook With CD-ROM PDFUyanga Natsagdorj83% (12)
- Phylum PoriferaDocument30 pagesPhylum PoriferaEloisa Lopez100% (2)
- Cell Management (5G RAN2.1 - Draft A)Document35 pagesCell Management (5G RAN2.1 - Draft A)Mohammed Mokhtar100% (9)
- TeachersBook EnglishFile Pre IntermediateDocument16 pagesTeachersBook EnglishFile Pre IntermediateBëkérzz Ãbîî JhîömêrNo ratings yet
- Dheeraj BishtDocument38 pagesDheeraj Bishtdheeraj bishtNo ratings yet
- 4 Gene ExpressionDocument35 pages4 Gene ExpressionThảo ThảoNo ratings yet
- Practical XII TH STD Zoology EMDocument24 pagesPractical XII TH STD Zoology EMsanthoshsivanandam SivanandamNo ratings yet
- BorreliaDocument19 pagesBorreliaZetti Lenon Vallejos GomezNo ratings yet
- Comparative Vertebrate Anatomy Lecture Notes - Digestive SystemDocument14 pagesComparative Vertebrate Anatomy Lecture Notes - Digestive SystemMielah Ruth100% (1)
- Transcription: RNA Polymerases and General Transcription FactorsDocument77 pagesTranscription: RNA Polymerases and General Transcription FactorsmluluNo ratings yet
- Ento231 PDFDocument151 pagesEnto231 PDFMunna GuptaNo ratings yet
- Locomotion ZoologyDocument12 pagesLocomotion Zoologyadithya4rajNo ratings yet
- Translation in Prokaryotes: NEET 2020Document1 pageTranslation in Prokaryotes: NEET 2020ADIKKI ANOOHYANo ratings yet
- Treatment of Pediculosis Capitis - A Critical Appraisal of The Current LiteratureDocument12 pagesTreatment of Pediculosis Capitis - A Critical Appraisal of The Current Literaturejft842No ratings yet
- 4 Porifera Spicules & Canal SystemDocument34 pages4 Porifera Spicules & Canal Systemapi-3732735100% (2)
- Farooq Ali: Presented byDocument22 pagesFarooq Ali: Presented byFarooq AliNo ratings yet
- Respiratory SurfacesDocument11 pagesRespiratory SurfacesNitesh KotianNo ratings yet
- Gifford-Gonzales ZOOARCHAEOLOGY An IntroductionDocument65 pagesGifford-Gonzales ZOOARCHAEOLOGY An IntroductionAleksandar TasićNo ratings yet
- Respiratory SystemDocument138 pagesRespiratory SystemJosefantastic 75No ratings yet
- Comparative Anatomy of Respiratory SystemDocument129 pagesComparative Anatomy of Respiratory Systemgerman guazaNo ratings yet
- Zoology Project of ZrsaDocument4 pagesZoology Project of ZrsaZotei RawihteNo ratings yet
- Wetlands of AssamDocument7 pagesWetlands of AssamHasibuddin AhmedNo ratings yet
- Composite Fish Culture in HillsDocument20 pagesComposite Fish Culture in HillsSullip MajhiNo ratings yet
- Eukaryote Regulation of Gene ExpressionDocument35 pagesEukaryote Regulation of Gene ExpressiondewiulfaNo ratings yet
- Human Digestive SystemDocument29 pagesHuman Digestive SystemRagini Dubey100% (1)
- NWIA National BrochureDocument32 pagesNWIA National BrochureSaurabh SinghviNo ratings yet
- Composite Fish Culture PDFDocument2 pagesComposite Fish Culture PDFHanumant JagtapNo ratings yet
- Cyclic AMP Signalling PathwayDocument5 pagesCyclic AMP Signalling Pathwayrawatpooran05No ratings yet
- Lec4 SocialInsects TheSociety NotesDocument3 pagesLec4 SocialInsects TheSociety NotesAlexandra LvNo ratings yet
- Pests of PaddyDocument19 pagesPests of PaddyPurushotham PaspuletiNo ratings yet
- 09 Vertebrate Digestive SystemDocument74 pages09 Vertebrate Digestive SystemJohn Andre CruzNo ratings yet
- Roles of Insects For HumanDocument58 pagesRoles of Insects For Humannovita manaluNo ratings yet
- ZOO301 Parade of VertebratesDocument14 pagesZOO301 Parade of VertebratesPatricia Hariramani100% (1)
- ZoologyDocument13 pagesZoologydvdmegaNo ratings yet
- Pre-mRNA: 5' Capping Polyadenylation Intron SplicingDocument1 pagePre-mRNA: 5' Capping Polyadenylation Intron SplicingKayoung HeoNo ratings yet
- Introduction To InsectsDocument7 pagesIntroduction To InsectsA R V KumarNo ratings yet
- Cnidaria SummaryDocument12 pagesCnidaria SummaryJose ArroyoNo ratings yet
- Archaeozoology: Camelopardalis Brisson, From The Prehistoric Site of Susunia, District BankuraDocument7 pagesArchaeozoology: Camelopardalis Brisson, From The Prehistoric Site of Susunia, District BankuraDibyendu DasNo ratings yet
- LiceDocument22 pagesLiceKalash NeupaneNo ratings yet
- Eukaryotic Dna Replication PDFDocument2 pagesEukaryotic Dna Replication PDFHughNo ratings yet
- Post Transcriptional ModificationDocument20 pagesPost Transcriptional ModificationZain YaqoobNo ratings yet
- Post Translational ModificationsDocument14 pagesPost Translational ModificationschainmaheNo ratings yet
- India Quiz QuestionsDocument12 pagesIndia Quiz Questionsankur0708No ratings yet
- Eukaryotic Protein Synthesis Differs From Prokaryotic Protein Synthesis Primarily in Translation InitiationDocument4 pagesEukaryotic Protein Synthesis Differs From Prokaryotic Protein Synthesis Primarily in Translation InitiationJoyce DometitaNo ratings yet
- Submitted by Aiswarya V 1St MSC Zoology Roll Number 3301Document32 pagesSubmitted by Aiswarya V 1St MSC Zoology Roll Number 3301PES PEOPLENo ratings yet
- Post Transcriptional ModificationDocument4 pagesPost Transcriptional Modificationrustyryan770% (1)
- Translational Control & Post-Translational ModificationsDocument10 pagesTranslational Control & Post-Translational ModificationsBhaskar GangulyNo ratings yet
- Management of Insect Pests of TobaccoDocument5 pagesManagement of Insect Pests of TobaccoStaidCasperNo ratings yet
- Dna Replication in EukaryotesDocument7 pagesDna Replication in EukaryotesAnish Shrestha100% (1)
- 1.pests of MulberryDocument28 pages1.pests of MulberrySomdip MojumdarNo ratings yet
- SYSTEMATICSDocument6 pagesSYSTEMATICSShelo Angielou Zurita100% (1)
- Genome Organization in ProkaryotesDocument8 pagesGenome Organization in ProkaryotesVijay Kishore75% (4)
- Gregor MendelDocument3 pagesGregor MendelAimee GutierrezNo ratings yet
- Translation in Prokaryotes and EukaryotesDocument17 pagesTranslation in Prokaryotes and EukaryotesMuhammad Arslan YasinNo ratings yet
- Clasification of InsectsDocument68 pagesClasification of InsectsMarie Serena McConnellNo ratings yet
- Post Translational ModificationDocument11 pagesPost Translational ModificationCynCyntya HarlyanaNo ratings yet
- Bacterial TransposonsDocument36 pagesBacterial TransposonsShweta SinghNo ratings yet
- Bernays & Chapman 1994Document325 pagesBernays & Chapman 1994Alexandre Pimenta100% (2)
- Zoology 100 Notes 1Document8 pagesZoology 100 Notes 1Bethany Jane Ravelo IsidroNo ratings yet
- Freshwater Microbiology: Biodiversity and Dynamic Interactions of Microorganisms in the Aquatic EnvironmentFrom EverandFreshwater Microbiology: Biodiversity and Dynamic Interactions of Microorganisms in the Aquatic EnvironmentNo ratings yet
- Pedersen - Gem and Ornamental Materials of Organic OriginDocument283 pagesPedersen - Gem and Ornamental Materials of Organic OriginLeila MazzonNo ratings yet
- Defect Bash - Literature Review: July 2013Document8 pagesDefect Bash - Literature Review: July 2013Narayanan SNNo ratings yet
- Content: - Introduction - Types of Solar Collector - Application - Advantage - DisadvantageDocument32 pagesContent: - Introduction - Types of Solar Collector - Application - Advantage - DisadvantagesivaramakrishnanNo ratings yet
- Control Engineering Viva & Interview Questions: Ssasit, SuratDocument11 pagesControl Engineering Viva & Interview Questions: Ssasit, SuratMuskan JainNo ratings yet
- Adarsh Public School: C-Block, Vikas PuriDocument2 pagesAdarsh Public School: C-Block, Vikas PuriNirmal KishorNo ratings yet
- Tugas Bahasa InggrisDocument6 pagesTugas Bahasa InggrisASICK MOTORVLOGNo ratings yet
- Indischee Bereding) Di Kecamatan Kintamani, BangliDocument12 pagesIndischee Bereding) Di Kecamatan Kintamani, BangliRuth ElferawiNo ratings yet
- Organisational Structure and DesignDocument25 pagesOrganisational Structure and DesignBright MulengaNo ratings yet
- 9i How To Setup Oracle Streams Replication. (Doc ID 224255.1)Document9 pages9i How To Setup Oracle Streams Replication. (Doc ID 224255.1)Nick SantosNo ratings yet
- Angelo Galasso Takes Space at Plaza's Edwardian Room - Retail - Business - Men's Wear - WWDDocument2 pagesAngelo Galasso Takes Space at Plaza's Edwardian Room - Retail - Business - Men's Wear - WWDamir.workNo ratings yet
- HMT Milling Machine TRM 3VDocument2 pagesHMT Milling Machine TRM 3Vramu100% (1)
- ENG ป.4-6 ชุด2Document18 pagesENG ป.4-6 ชุด2Laksami ChaitrintawatNo ratings yet
- 9 Edition: FPSI's Test Preparation Manual (TPM) With PST Items For Entry-Level Firefighters Order FormDocument1 page9 Edition: FPSI's Test Preparation Manual (TPM) With PST Items For Entry-Level Firefighters Order Formdragunfire50No ratings yet
- 2 The Power of Positive ThinkingDocument15 pages2 The Power of Positive ThinkingMark Laurence GuillesNo ratings yet
- Mar 31 2020 Mar 31 2020: Shogun-Method-Derek-Rake 1/5 Shogun-Method-Derek-Rake 1/5Document5 pagesMar 31 2020 Mar 31 2020: Shogun-Method-Derek-Rake 1/5 Shogun-Method-Derek-Rake 1/5Mwila TumbaNo ratings yet
- UEFA Football Sustainability StrategyDocument33 pagesUEFA Football Sustainability StrategykoszicsaNo ratings yet
- Cheque Collection Policy: Page 1 of 10Document10 pagesCheque Collection Policy: Page 1 of 10PIYUSH ARORANo ratings yet
- Water Supply Problems and SolutionsDocument14 pagesWater Supply Problems and SolutionsNobodyNo ratings yet
- 2013 Wassce Gka-ObjectivesDocument8 pages2013 Wassce Gka-ObjectivesKwabena AgyepongNo ratings yet
- Project MusthafaDocument64 pagesProject MusthafaMusthafa CkNo ratings yet
- The Goldberg Tire Company Manufactures Racing Tires For Bicycles GoldbergDocument2 pagesThe Goldberg Tire Company Manufactures Racing Tires For Bicycles GoldbergAmit PandeyNo ratings yet
- Important Questions Physics CLASS 11Document4 pagesImportant Questions Physics CLASS 11Palak AgarwalNo ratings yet
- Leadership and Managing Excellence AssignmentDocument4 pagesLeadership and Managing Excellence AssignmentSahil GuptaNo ratings yet
- Language and Linguistics Solved MCQs (Set-1)Document6 pagesLanguage and Linguistics Solved MCQs (Set-1)Rai Zia Ur RahmanNo ratings yet
- Corporate BrochureDocument28 pagesCorporate BrochureVijay KambleNo ratings yet
- Cool The Summer - Leaflet - HRDocument48 pagesCool The Summer - Leaflet - HRPeter Ace John IngelNo ratings yet
























































































Download as pdf or txt
You might also like
- Merritt's BakeryDocument1 pageMerritt's BakeryNardine Farag0% (1)
- Cambridge IGCSE Chemistry Coursebook With CD-ROM PDFDocument532 pagesCambridge IGCSE Chemistry Coursebook With CD-ROM PDFUyanga Natsagdorj83% (12)
- Phylum PoriferaDocument30 pagesPhylum PoriferaEloisa Lopez100% (2)
- Cell Management (5G RAN2.1 - Draft A)Document35 pagesCell Management (5G RAN2.1 - Draft A)Mohammed Mokhtar100% (9)
- TeachersBook EnglishFile Pre IntermediateDocument16 pagesTeachersBook EnglishFile Pre IntermediateBëkérzz Ãbîî JhîömêrNo ratings yet
- Dheeraj BishtDocument38 pagesDheeraj Bishtdheeraj bishtNo ratings yet
- 4 Gene ExpressionDocument35 pages4 Gene ExpressionThảo ThảoNo ratings yet
- Practical XII TH STD Zoology EMDocument24 pagesPractical XII TH STD Zoology EMsanthoshsivanandam SivanandamNo ratings yet
- BorreliaDocument19 pagesBorreliaZetti Lenon Vallejos GomezNo ratings yet
- Comparative Vertebrate Anatomy Lecture Notes - Digestive SystemDocument14 pagesComparative Vertebrate Anatomy Lecture Notes - Digestive SystemMielah Ruth100% (1)
- Transcription: RNA Polymerases and General Transcription FactorsDocument77 pagesTranscription: RNA Polymerases and General Transcription FactorsmluluNo ratings yet
- Ento231 PDFDocument151 pagesEnto231 PDFMunna GuptaNo ratings yet
- Locomotion ZoologyDocument12 pagesLocomotion Zoologyadithya4rajNo ratings yet
- Translation in Prokaryotes: NEET 2020Document1 pageTranslation in Prokaryotes: NEET 2020ADIKKI ANOOHYANo ratings yet
- Treatment of Pediculosis Capitis - A Critical Appraisal of The Current LiteratureDocument12 pagesTreatment of Pediculosis Capitis - A Critical Appraisal of The Current Literaturejft842No ratings yet
- 4 Porifera Spicules & Canal SystemDocument34 pages4 Porifera Spicules & Canal Systemapi-3732735100% (2)
- Farooq Ali: Presented byDocument22 pagesFarooq Ali: Presented byFarooq AliNo ratings yet
- Respiratory SurfacesDocument11 pagesRespiratory SurfacesNitesh KotianNo ratings yet
- Gifford-Gonzales ZOOARCHAEOLOGY An IntroductionDocument65 pagesGifford-Gonzales ZOOARCHAEOLOGY An IntroductionAleksandar TasićNo ratings yet
- Respiratory SystemDocument138 pagesRespiratory SystemJosefantastic 75No ratings yet
- Comparative Anatomy of Respiratory SystemDocument129 pagesComparative Anatomy of Respiratory Systemgerman guazaNo ratings yet
- Zoology Project of ZrsaDocument4 pagesZoology Project of ZrsaZotei RawihteNo ratings yet
- Wetlands of AssamDocument7 pagesWetlands of AssamHasibuddin AhmedNo ratings yet
- Composite Fish Culture in HillsDocument20 pagesComposite Fish Culture in HillsSullip MajhiNo ratings yet
- Eukaryote Regulation of Gene ExpressionDocument35 pagesEukaryote Regulation of Gene ExpressiondewiulfaNo ratings yet
- Human Digestive SystemDocument29 pagesHuman Digestive SystemRagini Dubey100% (1)
- NWIA National BrochureDocument32 pagesNWIA National BrochureSaurabh SinghviNo ratings yet
- Composite Fish Culture PDFDocument2 pagesComposite Fish Culture PDFHanumant JagtapNo ratings yet
- Cyclic AMP Signalling PathwayDocument5 pagesCyclic AMP Signalling Pathwayrawatpooran05No ratings yet
- Lec4 SocialInsects TheSociety NotesDocument3 pagesLec4 SocialInsects TheSociety NotesAlexandra LvNo ratings yet
- Pests of PaddyDocument19 pagesPests of PaddyPurushotham PaspuletiNo ratings yet
- 09 Vertebrate Digestive SystemDocument74 pages09 Vertebrate Digestive SystemJohn Andre CruzNo ratings yet
- Roles of Insects For HumanDocument58 pagesRoles of Insects For Humannovita manaluNo ratings yet
- ZOO301 Parade of VertebratesDocument14 pagesZOO301 Parade of VertebratesPatricia Hariramani100% (1)
- ZoologyDocument13 pagesZoologydvdmegaNo ratings yet
- Pre-mRNA: 5' Capping Polyadenylation Intron SplicingDocument1 pagePre-mRNA: 5' Capping Polyadenylation Intron SplicingKayoung HeoNo ratings yet
- Introduction To InsectsDocument7 pagesIntroduction To InsectsA R V KumarNo ratings yet
- Cnidaria SummaryDocument12 pagesCnidaria SummaryJose ArroyoNo ratings yet
- Archaeozoology: Camelopardalis Brisson, From The Prehistoric Site of Susunia, District BankuraDocument7 pagesArchaeozoology: Camelopardalis Brisson, From The Prehistoric Site of Susunia, District BankuraDibyendu DasNo ratings yet
- LiceDocument22 pagesLiceKalash NeupaneNo ratings yet
- Eukaryotic Dna Replication PDFDocument2 pagesEukaryotic Dna Replication PDFHughNo ratings yet
- Post Transcriptional ModificationDocument20 pagesPost Transcriptional ModificationZain YaqoobNo ratings yet
- Post Translational ModificationsDocument14 pagesPost Translational ModificationschainmaheNo ratings yet
- India Quiz QuestionsDocument12 pagesIndia Quiz Questionsankur0708No ratings yet
- Eukaryotic Protein Synthesis Differs From Prokaryotic Protein Synthesis Primarily in Translation InitiationDocument4 pagesEukaryotic Protein Synthesis Differs From Prokaryotic Protein Synthesis Primarily in Translation InitiationJoyce DometitaNo ratings yet
- Submitted by Aiswarya V 1St MSC Zoology Roll Number 3301Document32 pagesSubmitted by Aiswarya V 1St MSC Zoology Roll Number 3301PES PEOPLENo ratings yet
- Post Transcriptional ModificationDocument4 pagesPost Transcriptional Modificationrustyryan770% (1)
- Translational Control & Post-Translational ModificationsDocument10 pagesTranslational Control & Post-Translational ModificationsBhaskar GangulyNo ratings yet
- Management of Insect Pests of TobaccoDocument5 pagesManagement of Insect Pests of TobaccoStaidCasperNo ratings yet
- Dna Replication in EukaryotesDocument7 pagesDna Replication in EukaryotesAnish Shrestha100% (1)
- 1.pests of MulberryDocument28 pages1.pests of MulberrySomdip MojumdarNo ratings yet
- SYSTEMATICSDocument6 pagesSYSTEMATICSShelo Angielou Zurita100% (1)
- Genome Organization in ProkaryotesDocument8 pagesGenome Organization in ProkaryotesVijay Kishore75% (4)
- Gregor MendelDocument3 pagesGregor MendelAimee GutierrezNo ratings yet
- Translation in Prokaryotes and EukaryotesDocument17 pagesTranslation in Prokaryotes and EukaryotesMuhammad Arslan YasinNo ratings yet
- Clasification of InsectsDocument68 pagesClasification of InsectsMarie Serena McConnellNo ratings yet
- Post Translational ModificationDocument11 pagesPost Translational ModificationCynCyntya HarlyanaNo ratings yet
- Bacterial TransposonsDocument36 pagesBacterial TransposonsShweta SinghNo ratings yet
- Bernays & Chapman 1994Document325 pagesBernays & Chapman 1994Alexandre Pimenta100% (2)
- Zoology 100 Notes 1Document8 pagesZoology 100 Notes 1Bethany Jane Ravelo IsidroNo ratings yet
- Freshwater Microbiology: Biodiversity and Dynamic Interactions of Microorganisms in the Aquatic EnvironmentFrom EverandFreshwater Microbiology: Biodiversity and Dynamic Interactions of Microorganisms in the Aquatic EnvironmentNo ratings yet
- Pedersen - Gem and Ornamental Materials of Organic OriginDocument283 pagesPedersen - Gem and Ornamental Materials of Organic OriginLeila MazzonNo ratings yet
- Defect Bash - Literature Review: July 2013Document8 pagesDefect Bash - Literature Review: July 2013Narayanan SNNo ratings yet
- Content: - Introduction - Types of Solar Collector - Application - Advantage - DisadvantageDocument32 pagesContent: - Introduction - Types of Solar Collector - Application - Advantage - DisadvantagesivaramakrishnanNo ratings yet
- Control Engineering Viva & Interview Questions: Ssasit, SuratDocument11 pagesControl Engineering Viva & Interview Questions: Ssasit, SuratMuskan JainNo ratings yet
- Adarsh Public School: C-Block, Vikas PuriDocument2 pagesAdarsh Public School: C-Block, Vikas PuriNirmal KishorNo ratings yet
- Tugas Bahasa InggrisDocument6 pagesTugas Bahasa InggrisASICK MOTORVLOGNo ratings yet
- Indischee Bereding) Di Kecamatan Kintamani, BangliDocument12 pagesIndischee Bereding) Di Kecamatan Kintamani, BangliRuth ElferawiNo ratings yet
- Organisational Structure and DesignDocument25 pagesOrganisational Structure and DesignBright MulengaNo ratings yet
- 9i How To Setup Oracle Streams Replication. (Doc ID 224255.1)Document9 pages9i How To Setup Oracle Streams Replication. (Doc ID 224255.1)Nick SantosNo ratings yet
- Angelo Galasso Takes Space at Plaza's Edwardian Room - Retail - Business - Men's Wear - WWDDocument2 pagesAngelo Galasso Takes Space at Plaza's Edwardian Room - Retail - Business - Men's Wear - WWDamir.workNo ratings yet
- HMT Milling Machine TRM 3VDocument2 pagesHMT Milling Machine TRM 3Vramu100% (1)
- ENG ป.4-6 ชุด2Document18 pagesENG ป.4-6 ชุด2Laksami ChaitrintawatNo ratings yet
- 9 Edition: FPSI's Test Preparation Manual (TPM) With PST Items For Entry-Level Firefighters Order FormDocument1 page9 Edition: FPSI's Test Preparation Manual (TPM) With PST Items For Entry-Level Firefighters Order Formdragunfire50No ratings yet
- 2 The Power of Positive ThinkingDocument15 pages2 The Power of Positive ThinkingMark Laurence GuillesNo ratings yet
- Mar 31 2020 Mar 31 2020: Shogun-Method-Derek-Rake 1/5 Shogun-Method-Derek-Rake 1/5Document5 pagesMar 31 2020 Mar 31 2020: Shogun-Method-Derek-Rake 1/5 Shogun-Method-Derek-Rake 1/5Mwila TumbaNo ratings yet
- UEFA Football Sustainability StrategyDocument33 pagesUEFA Football Sustainability StrategykoszicsaNo ratings yet
- Cheque Collection Policy: Page 1 of 10Document10 pagesCheque Collection Policy: Page 1 of 10PIYUSH ARORANo ratings yet
- Water Supply Problems and SolutionsDocument14 pagesWater Supply Problems and SolutionsNobodyNo ratings yet
- 2013 Wassce Gka-ObjectivesDocument8 pages2013 Wassce Gka-ObjectivesKwabena AgyepongNo ratings yet
- Project MusthafaDocument64 pagesProject MusthafaMusthafa CkNo ratings yet
- The Goldberg Tire Company Manufactures Racing Tires For Bicycles GoldbergDocument2 pagesThe Goldberg Tire Company Manufactures Racing Tires For Bicycles GoldbergAmit PandeyNo ratings yet
- Important Questions Physics CLASS 11Document4 pagesImportant Questions Physics CLASS 11Palak AgarwalNo ratings yet
- Leadership and Managing Excellence AssignmentDocument4 pagesLeadership and Managing Excellence AssignmentSahil GuptaNo ratings yet
- Language and Linguistics Solved MCQs (Set-1)Document6 pagesLanguage and Linguistics Solved MCQs (Set-1)Rai Zia Ur RahmanNo ratings yet
- Corporate BrochureDocument28 pagesCorporate BrochureVijay KambleNo ratings yet
- Cool The Summer - Leaflet - HRDocument48 pagesCool The Summer - Leaflet - HRPeter Ace John IngelNo ratings yet