
Antituberculosis Drugs
Antituberculosis Drugs
You might also like
- Astm C 423Document12 pagesAstm C 423Abhinav AcharyaNo ratings yet
- Pricing Your Work Corporate and Industrial PhotographyDocument15 pagesPricing Your Work Corporate and Industrial PhotographyManu Mnau MnauNo ratings yet
- The Quest For The Holy Grail: New Antitubercular Chemical Entities, Targets and StrategiesDocument9 pagesThe Quest For The Holy Grail: New Antitubercular Chemical Entities, Targets and StrategiesMehari AsratNo ratings yet
- 4.targeting DNA Replication and Repair For The Development of NovelDocument18 pages4.targeting DNA Replication and Repair For The Development of NovelfeNo ratings yet
- Urgency of Novel Anti-Tuberculosis Strategies: A Prospective ChallengeDocument16 pagesUrgency of Novel Anti-Tuberculosis Strategies: A Prospective ChallengeArshia NazirNo ratings yet
- Current Research in Pharmacology and Drug DiscoveryDocument10 pagesCurrent Research in Pharmacology and Drug Discoverydev darma karinggaNo ratings yet
- Tuberculosis:Development of New Drugs and Treatment RegimensDocument10 pagesTuberculosis:Development of New Drugs and Treatment RegimensMedylin DualloNo ratings yet
- Evaluating Bedaquiline As A Treatment Option For Multidrug-Resistant TuberculosisDocument8 pagesEvaluating Bedaquiline As A Treatment Option For Multidrug-Resistant Tuberculosisa29921No ratings yet
- Template Artikel - Heryanti S2 Ubaya - PretomanidDocument11 pagesTemplate Artikel - Heryanti S2 Ubaya - PretomanidHeryanti PusparisaNo ratings yet
- Be Daqui LinaDocument10 pagesBe Daqui LinaMario Jimenez JimenezNo ratings yet
- What Is New in Management of Pediatric TuberculosisDocument9 pagesWhat Is New in Management of Pediatric Tuberculosisviani ayalNo ratings yet
- Ijms-22-00735 Id enDocument28 pagesIjms-22-00735 Id enNur AnisaNo ratings yet
- Assistant Professor, Department of Pharmacology, KLEU's JN Medical College, Belgaum - Karnataka. IndiaDocument5 pagesAssistant Professor, Department of Pharmacology, KLEU's JN Medical College, Belgaum - Karnataka. IndiaMahesh T MadhavanNo ratings yet
- Treatment Outcomes Among Pediatric Patients With Highly Drug-Resistant Tuberculosis, The Role of New and Repurposed Second-Line Tuberculosis DrugsDocument11 pagesTreatment Outcomes Among Pediatric Patients With Highly Drug-Resistant Tuberculosis, The Role of New and Repurposed Second-Line Tuberculosis DrugsKang LimbaNo ratings yet
- TRT2017 4920209Document12 pagesTRT2017 4920209ADVOCATE ASHUTOSH SHARMANo ratings yet
- Ginsberg and Spigelman Nat Med 2007Document5 pagesGinsberg and Spigelman Nat Med 2007hst939No ratings yet
- 5 RaDocument19 pages5 RaNitish TankNo ratings yet
- Zhang 2015Document15 pagesZhang 2015Annisa Nurfiatul AiniNo ratings yet
- Anti TBdrugsDocument22 pagesAnti TBdrugs118 Jenis VoraNo ratings yet
- JurnalDocument4 pagesJurnalAnonymous pfHZusnNo ratings yet
- DKX 506Document14 pagesDKX 506د. محمد سند راشد الحصينيNo ratings yet
- 16 RaDocument10 pages16 RaNitish TankNo ratings yet
- Minireview: New Drugs Against Tuberculosis: Problems, Progress, and Evaluation of Agents in Clinical DevelopmentDocument14 pagesMinireview: New Drugs Against Tuberculosis: Problems, Progress, and Evaluation of Agents in Clinical DevelopmentcgandarelaNo ratings yet
- Research Paper On MDR TBDocument6 pagesResearch Paper On MDR TBgz8qs4dn100% (1)
- Nano-Based Anti-Tubercular Drug Delivery: An Emerging Paradigm For Improved Therapeutic InterventionDocument11 pagesNano-Based Anti-Tubercular Drug Delivery: An Emerging Paradigm For Improved Therapeutic InterventionADVOCATE ASHUTOSH SHARMANo ratings yet
- Pharmacological Research: ReviewDocument12 pagesPharmacological Research: ReviewHeryanti PusparisaNo ratings yet
- BMJ m216 FullDocument18 pagesBMJ m216 Fulldiaspalma.medNo ratings yet
- TUBERCULOSISDocument4 pagesTUBERCULOSISLâm NguyễnNo ratings yet
- Policy Guidance On Drug-Susceptibility Testing (DST) of Second-Line Antituberculosis DrugsDocument20 pagesPolicy Guidance On Drug-Susceptibility Testing (DST) of Second-Line Antituberculosis DrugsrehanaNo ratings yet
- International Journal of Infectious Diseases: SciencedirectDocument5 pagesInternational Journal of Infectious Diseases: Sciencedirectbe a doctor for you Medical studentNo ratings yet
- Art 00004Document6 pagesArt 00004rperdinandiNo ratings yet
- Jurnal Korea Pemilihan Regimen Long Dari Pada Regimen ShortDocument9 pagesJurnal Korea Pemilihan Regimen Long Dari Pada Regimen Shorthasan andrianNo ratings yet
- Anti-Infectives Drug Advisory Committee MeetingDocument253 pagesAnti-Infectives Drug Advisory Committee MeetingbellamajNo ratings yet
- Burden of Tuberculosis - Combating Drug Resistance: EditorialDocument3 pagesBurden of Tuberculosis - Combating Drug Resistance: EditorialmominamalikNo ratings yet
- (MDR-TB) Di Indonesia: Tinjauan Sistematik: Faktor Keberhasilan Pengobatan Multi Drug Resistance TuberculosaDocument7 pages(MDR-TB) Di Indonesia: Tinjauan Sistematik: Faktor Keberhasilan Pengobatan Multi Drug Resistance TuberculosaAngieNo ratings yet
- Diagnosis and Treatment of Multidrug-Resistant TuberculosisDocument12 pagesDiagnosis and Treatment of Multidrug-Resistant Tuberculosisfeby megaNo ratings yet
- Jurnal MDR TBDocument7 pagesJurnal MDR TBIndra HebatNo ratings yet
- Cegielski, Levo Vs Moxi 3Document13 pagesCegielski, Levo Vs Moxi 3hasan andrianNo ratings yet
- MultidrugDocument3 pagesMultidrugMelisa MalikNo ratings yet
- Refrat TBDocument15 pagesRefrat TBAndika TatagNo ratings yet
- multi drug resistance in childrenDocument11 pagesmulti drug resistance in childrenMuneeb AltafNo ratings yet
- 360 1442 2 PBDocument8 pages360 1442 2 PBEghar EverydayishellNo ratings yet
- 2010.... Multitargeting OmpoundDocument17 pages2010.... Multitargeting OmpoundVAISHALI PATELNo ratings yet
- JMC2013 Development of Antituberculosis AgentsDocument6 pagesJMC2013 Development of Antituberculosis AgentsVincent GeruszNo ratings yet
- 1 s2.0 S0753332220312968 MainDocument11 pages1 s2.0 S0753332220312968 MainCristina SorairesNo ratings yet
- Bedaquiline, Delamanid, Linezolid, and Clofazimine For Treatment of Pre-Extensively Drug-Resistant Tuberculosis 2022Document9 pagesBedaquiline, Delamanid, Linezolid, and Clofazimine For Treatment of Pre-Extensively Drug-Resistant Tuberculosis 2022awaluddin andyNo ratings yet
- Development of New Drugs For TB Chemotherapy: Analysis of The Current Drug PipelineDocument48 pagesDevelopment of New Drugs For TB Chemotherapy: Analysis of The Current Drug Pipelinereadyboy89No ratings yet
- Palliative and End-Of-Life Care in The Global Response To Multidrug-Resistant TuberculosisDocument7 pagesPalliative and End-Of-Life Care in The Global Response To Multidrug-Resistant TuberculosisSausan ZakiyahNo ratings yet
- Drug Resistant Tuberculosis Advances in Diagnosis.9Document7 pagesDrug Resistant Tuberculosis Advances in Diagnosis.9Victor Akinseye OluwatoyinNo ratings yet
- WHO Bulletin 2005Document10 pagesWHO Bulletin 2005ajh2675No ratings yet
- American Thoracic Society Documents: Treatment of Drug-Resistant TuberculosisDocument11 pagesAmerican Thoracic Society Documents: Treatment of Drug-Resistant TuberculosisanisaNo ratings yet
- Research Proposal - Lamisa Nur - ID 201406080 - 4th Year 8th SemesterDocument12 pagesResearch Proposal - Lamisa Nur - ID 201406080 - 4th Year 8th Semesteriksumaiya29No ratings yet
- Adverse Drug Reactions and Outcome Analysis of MDR TB Patients On Dots Plus RegimenDocument5 pagesAdverse Drug Reactions and Outcome Analysis of MDR TB Patients On Dots Plus RegimenkopaljsNo ratings yet
- No 2Document6 pagesNo 2Raven RavenNo ratings yet
- Eficacy and Safety Rejimen Long Vs Rejimen Short in TB MDRDocument12 pagesEficacy and Safety Rejimen Long Vs Rejimen Short in TB MDRhasan andrianNo ratings yet
- First-Line Anti-Tuberculosis Drug Resistance Pattern: Original ArticleDocument6 pagesFirst-Line Anti-Tuberculosis Drug Resistance Pattern: Original ArticleyasserNo ratings yet
- A Long-Acting Formulation of Rifabutin Is Effective For Prevention and Treatment of Mycobacterium TuberculosisDocument16 pagesA Long-Acting Formulation of Rifabutin Is Effective For Prevention and Treatment of Mycobacterium TuberculosisAyu RahayuNo ratings yet
- AMR M Tuberculosis PerspectiveDocument20 pagesAMR M Tuberculosis Perspectiveyves.cosnuauNo ratings yet
- Incidence and Predictors of Adverse Drug Events Among People Receiving Drug Resistant Tuberculosis Treatment in Uganda 8-Year Retrospective Cohort STDocument12 pagesIncidence and Predictors of Adverse Drug Events Among People Receiving Drug Resistant Tuberculosis Treatment in Uganda 8-Year Retrospective Cohort STEghar EverydayishellNo ratings yet
- Review-Pipeline of Anti-Mycobacterium Abscessus Small MoleculesDocument38 pagesReview-Pipeline of Anti-Mycobacterium Abscessus Small MoleculesMayuriNo ratings yet
- Singh., 2020. Mekanisme Resistensi ObatDocument21 pagesSingh., 2020. Mekanisme Resistensi Obathasan andrianNo ratings yet
- Critical Appraisal of Clinical ResearchDocument9 pagesCritical Appraisal of Clinical ResearchI Kadek Adi Putra Suandana 2005No ratings yet
- Stability Comparison of Tulasi (Ocimum Tenuiflorum L.) Leaf Gel Hand Sanitizer Using 0.5% and 1% Cmc-NaDocument2 pagesStability Comparison of Tulasi (Ocimum Tenuiflorum L.) Leaf Gel Hand Sanitizer Using 0.5% and 1% Cmc-NaI Kadek Adi Putra Suandana 2005No ratings yet
- English - Reading A Scientific PaperDocument23 pagesEnglish - Reading A Scientific PaperI Kadek Adi Putra Suandana 2005No ratings yet
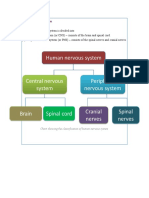
- Human Nervous SystemDocument6 pagesHuman Nervous SystemI Kadek Adi Putra Suandana 2005No ratings yet
- 10 Star FarmasiDocument3 pages10 Star FarmasiI Kadek Adi Putra Suandana 2005No ratings yet
- English Task 3Document6 pagesEnglish Task 3I Kadek Adi Putra Suandana 2005No ratings yet
- 10 Star FarmasiDocument3 pages10 Star FarmasiI Kadek Adi Putra Suandana 2005No ratings yet
- English Task 1 - 2008551005 - I Kadek Adi Putra SuandanaDocument1 pageEnglish Task 1 - 2008551005 - I Kadek Adi Putra SuandanaI Kadek Adi Putra Suandana 2005No ratings yet
- Cognizant Test 1Document20 pagesCognizant Test 1Veeraragavan SubramaniamNo ratings yet
- Current ElectricityDocument93 pagesCurrent ElectricitySurya SNo ratings yet
- Teacher: Diana Marie V. Aman Science Teaching Dates/Time: Quarter: SecondDocument6 pagesTeacher: Diana Marie V. Aman Science Teaching Dates/Time: Quarter: SecondDiana Marie Vidallon AmanNo ratings yet
- US Gasification DatabaseDocument9 pagesUS Gasification DatabaseAhmad DaoodNo ratings yet
- Kepler Problem - Wikipedia, The Free Encyclopedia PDFDocument4 pagesKepler Problem - Wikipedia, The Free Encyclopedia PDFrizal123No ratings yet
- 3) 2013 March - The Thing About Ms. Hollis MaynellDocument23 pages3) 2013 March - The Thing About Ms. Hollis MaynellShy TolentinoNo ratings yet
- ManualeDelphi IngleseDocument86 pagesManualeDelphi IngleseoxooxooxoNo ratings yet
- Common Prospectus EnglishDocument293 pagesCommon Prospectus Englishven23No ratings yet
- XXX (Topic) 好处影响1 相反好处 2. 你⾃自⼰己的观点,后⾯面会展开的Document3 pagesXXX (Topic) 好处影响1 相反好处 2. 你⾃自⼰己的观点,后⾯面会展开的Miyou KwanNo ratings yet
- Mathematics Past Paper QuestionsDocument174 pagesMathematics Past Paper Questionsnodicoh572100% (2)
- Biochemistry FinalDocument12 pagesBiochemistry FinalAhmed Hamarneh100% (1)
- Chap 09Document132 pagesChap 09noscribdyoucantNo ratings yet
- Biology: NO Judul PengarangDocument5 pagesBiology: NO Judul Pengarangkartini11No ratings yet
- SummaryDocument2 pagesSummaryRosida IdaNo ratings yet
- Lesson Plan 8 (September) (AutoRecovered) 1Document3 pagesLesson Plan 8 (September) (AutoRecovered) 1Rutchie AbantoNo ratings yet
- Infosys-Broadcom E2E Continuous Testing Platform Business Process Automation SolutionDocument16 pagesInfosys-Broadcom E2E Continuous Testing Platform Business Process Automation Solutioncharu.hitechrobot2889No ratings yet
- CEA Farms Logistics USA Slide Deck (2023!10!31 10-38-56 UTC)Document18 pagesCEA Farms Logistics USA Slide Deck (2023!10!31 10-38-56 UTC)SiyabongaNo ratings yet
- In-Band Full-Duplex Interference For Underwater Acoustic Communication SystemsDocument6 pagesIn-Band Full-Duplex Interference For Underwater Acoustic Communication SystemsHarris TsimenidisNo ratings yet
- HB-1193-006 HB PlasmidPurif 0723 WWDocument68 pagesHB-1193-006 HB PlasmidPurif 0723 WWDiana DiasNo ratings yet
- Biogas ProductionDocument7 pagesBiogas ProductionFagbohungbe MichaelNo ratings yet
- Comfort ZoneDocument4 pagesComfort Zonesigal ardanNo ratings yet
- Digital Banking A Case Study of India: Solid State Technology December 2020Document10 pagesDigital Banking A Case Study of India: Solid State Technology December 2020Mohit PacharNo ratings yet
- Sony Kdl-40xbr9 Kdl-46xbr9 Kdl-52xbr9 Ex2m ChassisDocument118 pagesSony Kdl-40xbr9 Kdl-46xbr9 Kdl-52xbr9 Ex2m ChassisAndy WilsonNo ratings yet
- VP3401 As04Document2 pagesVP3401 As04shivamtyagi68637No ratings yet
- HUAWEI - SUN2000-20-40KTL-M3-UserManual GCADocument102 pagesHUAWEI - SUN2000-20-40KTL-M3-UserManual GCAReardon MetalsNo ratings yet
- Taylor Swift LyricsDocument2 pagesTaylor Swift LyricsElsie DomeNo ratings yet
- English in Common 2b Split Student Book With Activebook and Workbook Volume 2 Part 2Document26 pagesEnglish in Common 2b Split Student Book With Activebook and Workbook Volume 2 Part 2Pancho NohalesNo ratings yet
- Tuyển Sinh 10 - đề 1 -KeyDocument5 pagesTuyển Sinh 10 - đề 1 -Keynguyenhoang17042004No ratings yet





























































































Download as pdf or txt
You might also like
- Astm C 423Document12 pagesAstm C 423Abhinav AcharyaNo ratings yet
- Pricing Your Work Corporate and Industrial PhotographyDocument15 pagesPricing Your Work Corporate and Industrial PhotographyManu Mnau MnauNo ratings yet
- The Quest For The Holy Grail: New Antitubercular Chemical Entities, Targets and StrategiesDocument9 pagesThe Quest For The Holy Grail: New Antitubercular Chemical Entities, Targets and StrategiesMehari AsratNo ratings yet
- 4.targeting DNA Replication and Repair For The Development of NovelDocument18 pages4.targeting DNA Replication and Repair For The Development of NovelfeNo ratings yet
- Urgency of Novel Anti-Tuberculosis Strategies: A Prospective ChallengeDocument16 pagesUrgency of Novel Anti-Tuberculosis Strategies: A Prospective ChallengeArshia NazirNo ratings yet
- Current Research in Pharmacology and Drug DiscoveryDocument10 pagesCurrent Research in Pharmacology and Drug Discoverydev darma karinggaNo ratings yet
- Tuberculosis:Development of New Drugs and Treatment RegimensDocument10 pagesTuberculosis:Development of New Drugs and Treatment RegimensMedylin DualloNo ratings yet
- Evaluating Bedaquiline As A Treatment Option For Multidrug-Resistant TuberculosisDocument8 pagesEvaluating Bedaquiline As A Treatment Option For Multidrug-Resistant Tuberculosisa29921No ratings yet
- Template Artikel - Heryanti S2 Ubaya - PretomanidDocument11 pagesTemplate Artikel - Heryanti S2 Ubaya - PretomanidHeryanti PusparisaNo ratings yet
- Be Daqui LinaDocument10 pagesBe Daqui LinaMario Jimenez JimenezNo ratings yet
- What Is New in Management of Pediatric TuberculosisDocument9 pagesWhat Is New in Management of Pediatric Tuberculosisviani ayalNo ratings yet
- Ijms-22-00735 Id enDocument28 pagesIjms-22-00735 Id enNur AnisaNo ratings yet
- Assistant Professor, Department of Pharmacology, KLEU's JN Medical College, Belgaum - Karnataka. IndiaDocument5 pagesAssistant Professor, Department of Pharmacology, KLEU's JN Medical College, Belgaum - Karnataka. IndiaMahesh T MadhavanNo ratings yet
- Treatment Outcomes Among Pediatric Patients With Highly Drug-Resistant Tuberculosis, The Role of New and Repurposed Second-Line Tuberculosis DrugsDocument11 pagesTreatment Outcomes Among Pediatric Patients With Highly Drug-Resistant Tuberculosis, The Role of New and Repurposed Second-Line Tuberculosis DrugsKang LimbaNo ratings yet
- TRT2017 4920209Document12 pagesTRT2017 4920209ADVOCATE ASHUTOSH SHARMANo ratings yet
- Ginsberg and Spigelman Nat Med 2007Document5 pagesGinsberg and Spigelman Nat Med 2007hst939No ratings yet
- 5 RaDocument19 pages5 RaNitish TankNo ratings yet
- Zhang 2015Document15 pagesZhang 2015Annisa Nurfiatul AiniNo ratings yet
- Anti TBdrugsDocument22 pagesAnti TBdrugs118 Jenis VoraNo ratings yet
- JurnalDocument4 pagesJurnalAnonymous pfHZusnNo ratings yet
- DKX 506Document14 pagesDKX 506د. محمد سند راشد الحصينيNo ratings yet
- 16 RaDocument10 pages16 RaNitish TankNo ratings yet
- Minireview: New Drugs Against Tuberculosis: Problems, Progress, and Evaluation of Agents in Clinical DevelopmentDocument14 pagesMinireview: New Drugs Against Tuberculosis: Problems, Progress, and Evaluation of Agents in Clinical DevelopmentcgandarelaNo ratings yet
- Research Paper On MDR TBDocument6 pagesResearch Paper On MDR TBgz8qs4dn100% (1)
- Nano-Based Anti-Tubercular Drug Delivery: An Emerging Paradigm For Improved Therapeutic InterventionDocument11 pagesNano-Based Anti-Tubercular Drug Delivery: An Emerging Paradigm For Improved Therapeutic InterventionADVOCATE ASHUTOSH SHARMANo ratings yet
- Pharmacological Research: ReviewDocument12 pagesPharmacological Research: ReviewHeryanti PusparisaNo ratings yet
- BMJ m216 FullDocument18 pagesBMJ m216 Fulldiaspalma.medNo ratings yet
- TUBERCULOSISDocument4 pagesTUBERCULOSISLâm NguyễnNo ratings yet
- Policy Guidance On Drug-Susceptibility Testing (DST) of Second-Line Antituberculosis DrugsDocument20 pagesPolicy Guidance On Drug-Susceptibility Testing (DST) of Second-Line Antituberculosis DrugsrehanaNo ratings yet
- International Journal of Infectious Diseases: SciencedirectDocument5 pagesInternational Journal of Infectious Diseases: Sciencedirectbe a doctor for you Medical studentNo ratings yet
- Art 00004Document6 pagesArt 00004rperdinandiNo ratings yet
- Jurnal Korea Pemilihan Regimen Long Dari Pada Regimen ShortDocument9 pagesJurnal Korea Pemilihan Regimen Long Dari Pada Regimen Shorthasan andrianNo ratings yet
- Anti-Infectives Drug Advisory Committee MeetingDocument253 pagesAnti-Infectives Drug Advisory Committee MeetingbellamajNo ratings yet
- Burden of Tuberculosis - Combating Drug Resistance: EditorialDocument3 pagesBurden of Tuberculosis - Combating Drug Resistance: EditorialmominamalikNo ratings yet
- (MDR-TB) Di Indonesia: Tinjauan Sistematik: Faktor Keberhasilan Pengobatan Multi Drug Resistance TuberculosaDocument7 pages(MDR-TB) Di Indonesia: Tinjauan Sistematik: Faktor Keberhasilan Pengobatan Multi Drug Resistance TuberculosaAngieNo ratings yet
- Diagnosis and Treatment of Multidrug-Resistant TuberculosisDocument12 pagesDiagnosis and Treatment of Multidrug-Resistant Tuberculosisfeby megaNo ratings yet
- Jurnal MDR TBDocument7 pagesJurnal MDR TBIndra HebatNo ratings yet
- Cegielski, Levo Vs Moxi 3Document13 pagesCegielski, Levo Vs Moxi 3hasan andrianNo ratings yet
- MultidrugDocument3 pagesMultidrugMelisa MalikNo ratings yet
- Refrat TBDocument15 pagesRefrat TBAndika TatagNo ratings yet
- multi drug resistance in childrenDocument11 pagesmulti drug resistance in childrenMuneeb AltafNo ratings yet
- 360 1442 2 PBDocument8 pages360 1442 2 PBEghar EverydayishellNo ratings yet
- 2010.... Multitargeting OmpoundDocument17 pages2010.... Multitargeting OmpoundVAISHALI PATELNo ratings yet
- JMC2013 Development of Antituberculosis AgentsDocument6 pagesJMC2013 Development of Antituberculosis AgentsVincent GeruszNo ratings yet
- 1 s2.0 S0753332220312968 MainDocument11 pages1 s2.0 S0753332220312968 MainCristina SorairesNo ratings yet
- Bedaquiline, Delamanid, Linezolid, and Clofazimine For Treatment of Pre-Extensively Drug-Resistant Tuberculosis 2022Document9 pagesBedaquiline, Delamanid, Linezolid, and Clofazimine For Treatment of Pre-Extensively Drug-Resistant Tuberculosis 2022awaluddin andyNo ratings yet
- Development of New Drugs For TB Chemotherapy: Analysis of The Current Drug PipelineDocument48 pagesDevelopment of New Drugs For TB Chemotherapy: Analysis of The Current Drug Pipelinereadyboy89No ratings yet
- Palliative and End-Of-Life Care in The Global Response To Multidrug-Resistant TuberculosisDocument7 pagesPalliative and End-Of-Life Care in The Global Response To Multidrug-Resistant TuberculosisSausan ZakiyahNo ratings yet
- Drug Resistant Tuberculosis Advances in Diagnosis.9Document7 pagesDrug Resistant Tuberculosis Advances in Diagnosis.9Victor Akinseye OluwatoyinNo ratings yet
- WHO Bulletin 2005Document10 pagesWHO Bulletin 2005ajh2675No ratings yet
- American Thoracic Society Documents: Treatment of Drug-Resistant TuberculosisDocument11 pagesAmerican Thoracic Society Documents: Treatment of Drug-Resistant TuberculosisanisaNo ratings yet
- Research Proposal - Lamisa Nur - ID 201406080 - 4th Year 8th SemesterDocument12 pagesResearch Proposal - Lamisa Nur - ID 201406080 - 4th Year 8th Semesteriksumaiya29No ratings yet
- Adverse Drug Reactions and Outcome Analysis of MDR TB Patients On Dots Plus RegimenDocument5 pagesAdverse Drug Reactions and Outcome Analysis of MDR TB Patients On Dots Plus RegimenkopaljsNo ratings yet
- No 2Document6 pagesNo 2Raven RavenNo ratings yet
- Eficacy and Safety Rejimen Long Vs Rejimen Short in TB MDRDocument12 pagesEficacy and Safety Rejimen Long Vs Rejimen Short in TB MDRhasan andrianNo ratings yet
- First-Line Anti-Tuberculosis Drug Resistance Pattern: Original ArticleDocument6 pagesFirst-Line Anti-Tuberculosis Drug Resistance Pattern: Original ArticleyasserNo ratings yet
- A Long-Acting Formulation of Rifabutin Is Effective For Prevention and Treatment of Mycobacterium TuberculosisDocument16 pagesA Long-Acting Formulation of Rifabutin Is Effective For Prevention and Treatment of Mycobacterium TuberculosisAyu RahayuNo ratings yet
- AMR M Tuberculosis PerspectiveDocument20 pagesAMR M Tuberculosis Perspectiveyves.cosnuauNo ratings yet
- Incidence and Predictors of Adverse Drug Events Among People Receiving Drug Resistant Tuberculosis Treatment in Uganda 8-Year Retrospective Cohort STDocument12 pagesIncidence and Predictors of Adverse Drug Events Among People Receiving Drug Resistant Tuberculosis Treatment in Uganda 8-Year Retrospective Cohort STEghar EverydayishellNo ratings yet
- Review-Pipeline of Anti-Mycobacterium Abscessus Small MoleculesDocument38 pagesReview-Pipeline of Anti-Mycobacterium Abscessus Small MoleculesMayuriNo ratings yet
- Singh., 2020. Mekanisme Resistensi ObatDocument21 pagesSingh., 2020. Mekanisme Resistensi Obathasan andrianNo ratings yet
- Critical Appraisal of Clinical ResearchDocument9 pagesCritical Appraisal of Clinical ResearchI Kadek Adi Putra Suandana 2005No ratings yet
- Stability Comparison of Tulasi (Ocimum Tenuiflorum L.) Leaf Gel Hand Sanitizer Using 0.5% and 1% Cmc-NaDocument2 pagesStability Comparison of Tulasi (Ocimum Tenuiflorum L.) Leaf Gel Hand Sanitizer Using 0.5% and 1% Cmc-NaI Kadek Adi Putra Suandana 2005No ratings yet
- English - Reading A Scientific PaperDocument23 pagesEnglish - Reading A Scientific PaperI Kadek Adi Putra Suandana 2005No ratings yet
- Human Nervous SystemDocument6 pagesHuman Nervous SystemI Kadek Adi Putra Suandana 2005No ratings yet
- 10 Star FarmasiDocument3 pages10 Star FarmasiI Kadek Adi Putra Suandana 2005No ratings yet
- English Task 3Document6 pagesEnglish Task 3I Kadek Adi Putra Suandana 2005No ratings yet
- 10 Star FarmasiDocument3 pages10 Star FarmasiI Kadek Adi Putra Suandana 2005No ratings yet
- English Task 1 - 2008551005 - I Kadek Adi Putra SuandanaDocument1 pageEnglish Task 1 - 2008551005 - I Kadek Adi Putra SuandanaI Kadek Adi Putra Suandana 2005No ratings yet
- Cognizant Test 1Document20 pagesCognizant Test 1Veeraragavan SubramaniamNo ratings yet
- Current ElectricityDocument93 pagesCurrent ElectricitySurya SNo ratings yet
- Teacher: Diana Marie V. Aman Science Teaching Dates/Time: Quarter: SecondDocument6 pagesTeacher: Diana Marie V. Aman Science Teaching Dates/Time: Quarter: SecondDiana Marie Vidallon AmanNo ratings yet
- US Gasification DatabaseDocument9 pagesUS Gasification DatabaseAhmad DaoodNo ratings yet
- Kepler Problem - Wikipedia, The Free Encyclopedia PDFDocument4 pagesKepler Problem - Wikipedia, The Free Encyclopedia PDFrizal123No ratings yet
- 3) 2013 March - The Thing About Ms. Hollis MaynellDocument23 pages3) 2013 March - The Thing About Ms. Hollis MaynellShy TolentinoNo ratings yet
- ManualeDelphi IngleseDocument86 pagesManualeDelphi IngleseoxooxooxoNo ratings yet
- Common Prospectus EnglishDocument293 pagesCommon Prospectus Englishven23No ratings yet
- XXX (Topic) 好处影响1 相反好处 2. 你⾃自⼰己的观点,后⾯面会展开的Document3 pagesXXX (Topic) 好处影响1 相反好处 2. 你⾃自⼰己的观点,后⾯面会展开的Miyou KwanNo ratings yet
- Mathematics Past Paper QuestionsDocument174 pagesMathematics Past Paper Questionsnodicoh572100% (2)
- Biochemistry FinalDocument12 pagesBiochemistry FinalAhmed Hamarneh100% (1)
- Chap 09Document132 pagesChap 09noscribdyoucantNo ratings yet
- Biology: NO Judul PengarangDocument5 pagesBiology: NO Judul Pengarangkartini11No ratings yet
- SummaryDocument2 pagesSummaryRosida IdaNo ratings yet
- Lesson Plan 8 (September) (AutoRecovered) 1Document3 pagesLesson Plan 8 (September) (AutoRecovered) 1Rutchie AbantoNo ratings yet
- Infosys-Broadcom E2E Continuous Testing Platform Business Process Automation SolutionDocument16 pagesInfosys-Broadcom E2E Continuous Testing Platform Business Process Automation Solutioncharu.hitechrobot2889No ratings yet
- CEA Farms Logistics USA Slide Deck (2023!10!31 10-38-56 UTC)Document18 pagesCEA Farms Logistics USA Slide Deck (2023!10!31 10-38-56 UTC)SiyabongaNo ratings yet
- In-Band Full-Duplex Interference For Underwater Acoustic Communication SystemsDocument6 pagesIn-Band Full-Duplex Interference For Underwater Acoustic Communication SystemsHarris TsimenidisNo ratings yet
- HB-1193-006 HB PlasmidPurif 0723 WWDocument68 pagesHB-1193-006 HB PlasmidPurif 0723 WWDiana DiasNo ratings yet
- Biogas ProductionDocument7 pagesBiogas ProductionFagbohungbe MichaelNo ratings yet
- Comfort ZoneDocument4 pagesComfort Zonesigal ardanNo ratings yet
- Digital Banking A Case Study of India: Solid State Technology December 2020Document10 pagesDigital Banking A Case Study of India: Solid State Technology December 2020Mohit PacharNo ratings yet
- Sony Kdl-40xbr9 Kdl-46xbr9 Kdl-52xbr9 Ex2m ChassisDocument118 pagesSony Kdl-40xbr9 Kdl-46xbr9 Kdl-52xbr9 Ex2m ChassisAndy WilsonNo ratings yet
- VP3401 As04Document2 pagesVP3401 As04shivamtyagi68637No ratings yet
- HUAWEI - SUN2000-20-40KTL-M3-UserManual GCADocument102 pagesHUAWEI - SUN2000-20-40KTL-M3-UserManual GCAReardon MetalsNo ratings yet
- Taylor Swift LyricsDocument2 pagesTaylor Swift LyricsElsie DomeNo ratings yet
- English in Common 2b Split Student Book With Activebook and Workbook Volume 2 Part 2Document26 pagesEnglish in Common 2b Split Student Book With Activebook and Workbook Volume 2 Part 2Pancho NohalesNo ratings yet
- Tuyển Sinh 10 - đề 1 -KeyDocument5 pagesTuyển Sinh 10 - đề 1 -Keynguyenhoang17042004No ratings yet