Download as pdf or txt
You might also like
- Performance Standards For Antifungal Susceptibility Testing of YeastsDocument30 pagesPerformance Standards For Antifungal Susceptibility Testing of YeastsAnne Abejuela Obial100% (2)
- Miracles From The Vault... More Than 90 Life Saving, Award Winning 'Underground Cures' (Health & Alternative Medicine) (Health Science Institute)Document6 pagesMiracles From The Vault... More Than 90 Life Saving, Award Winning 'Underground Cures' (Health & Alternative Medicine) (Health Science Institute)prakash0% (7)
- Caregiving Skill Evaluation TestDocument10 pagesCaregiving Skill Evaluation Testアビージョセフベンディホ ベロン100% (2)
- Endocrinology - Internal Medicine, Dr. A. Mowafy (2020-2021)Document172 pagesEndocrinology - Internal Medicine, Dr. A. Mowafy (2020-2021)Mohammed Risq100% (1)
- Inborn Errors of MetabolismDocument12 pagesInborn Errors of Metabolisma.engelherczNo ratings yet
- Jurnal Hormon 1Document7 pagesJurnal Hormon 12019072977No ratings yet
- Chapter 10 Assessment and Management of Patients With Endocrine DisordersDocument65 pagesChapter 10 Assessment and Management of Patients With Endocrine DisordersMYLENE GRACE ELARCOSA100% (2)
- 3.tumori Hipofizare 2018Document83 pages3.tumori Hipofizare 2018Oana AndreeaNo ratings yet
- Acromegaly and Gigantism: South Afr J Anaesth AnalgDocument5 pagesAcromegaly and Gigantism: South Afr J Anaesth AnalgatarNo ratings yet
- HyperpitutrismDocument41 pagesHyperpitutrismmuhammad kazimNo ratings yet
- Acromegalia Humanos X Gatos 2024Document11 pagesAcromegalia Humanos X Gatos 2024Lucas XavierNo ratings yet
- Alterations in Metabolism and Endocrine Problems HandoutsDocument15 pagesAlterations in Metabolism and Endocrine Problems HandoutsKeanu Win CatipayNo ratings yet
- Endocrine NotesDocument14 pagesEndocrine Notesrshannen19No ratings yet
- Androgen Insensitivity SyndromeDocument10 pagesAndrogen Insensitivity SyndromeAvino Mulana FikriNo ratings yet
- Pituitary Tumor & AcromegalyDocument32 pagesPituitary Tumor & Acromegalysam840720No ratings yet
- DR Atul B Mehta - Delay in Diagnosing Rare DiseasesDocument47 pagesDR Atul B Mehta - Delay in Diagnosing Rare Diseasesdanijara01No ratings yet
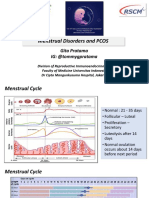
- Menstrual Disorders and PCOS Webinar EMBRYO FKUI 2022Document49 pagesMenstrual Disorders and PCOS Webinar EMBRYO FKUI 2022angelinputri100% (1)
- A 24-Year-Old Male With Gigantism, Growth Hormone Deficiency, Suspected Clivus Chordoma, Primary Hypothyroidism, Hypogonadism and PancytopeniaDocument8 pagesA 24-Year-Old Male With Gigantism, Growth Hormone Deficiency, Suspected Clivus Chordoma, Primary Hypothyroidism, Hypogonadism and PancytopeniaKiki SyamNo ratings yet
- Comprehensive Management of PCOS - GPDocument48 pagesComprehensive Management of PCOS - GPtata maretha100% (1)
- Congenital Adrenal Hyperplasia: SeminarDocument17 pagesCongenital Adrenal Hyperplasia: SeminarMaría Paula Sarmiento Ramón0% (1)
- Genetic Tests Can Help To:: 1. CytogeneticsDocument3 pagesGenetic Tests Can Help To:: 1. CytogeneticsRose Andrea De los Santos0% (1)
- Pituitary Gigantism: A Case Series From Hospital de San José (Bogotá, Colombia)Document9 pagesPituitary Gigantism: A Case Series From Hospital de San José (Bogotá, Colombia)Naila SalsaNo ratings yet
- B. 10 Situation - Care of Client With Problems in Metabolism & Endocrine Functioning.Document7 pagesB. 10 Situation - Care of Client With Problems in Metabolism & Endocrine Functioning.SOLEIL LOUISE LACSON MARBAS100% (1)
- 17-Mrdi DLP-1Document114 pages17-Mrdi DLP-1Jawad Ul HaqNo ratings yet
- GynecomastiaDocument22 pagesGynecomastiaIJAN DHAMALANo ratings yet
- PEDIA - Disorders of The Pituitary GlandDocument6 pagesPEDIA - Disorders of The Pituitary GlandEmerson QuimbaNo ratings yet
- Biopharmaceuticals Production PDFDocument29 pagesBiopharmaceuticals Production PDFEndra PratamaNo ratings yet
- 4.endocrine SummaryDocument143 pages4.endocrine SummaryAyatNo ratings yet
- Endocrinología Y Nutrición: Practical Guidelines For Diagnosis and Treatment of AcromegalyDocument15 pagesEndocrinología Y Nutrición: Practical Guidelines For Diagnosis and Treatment of Acromegalypancho gomezNo ratings yet
- Endocrine Passmedicine & Onexamination Notes 2016 PDFDocument96 pagesEndocrine Passmedicine & Onexamination Notes 2016 PDFsepan abdullahNo ratings yet
- Endocrine - Dr. Allam 2021Document69 pagesEndocrine - Dr. Allam 2021Alokh Saha RajNo ratings yet
- Erlotinib Monograph 1july2014Document5 pagesErlotinib Monograph 1july2014Wahyu RellyNo ratings yet
- NCM 107 Rle Nicu ResearchDocument3 pagesNCM 107 Rle Nicu ResearchIsha Catimbang GenerilloNo ratings yet
- R & C F T L: Eviews OmmentsDocument3 pagesR & C F T L: Eviews Ommentssanbin007No ratings yet
- AcromegalyDocument24 pagesAcromegalycsngiuNo ratings yet
- OBGYN RPL DR IndraDocument25 pagesOBGYN RPL DR Indraabraham winartoNo ratings yet
- Hypogonadism in MenDocument50 pagesHypogonadism in MenKate ClarksonNo ratings yet
- MTAPDocument19 pagesMTAPCaressa Marie EstradaNo ratings yet
- CAH Lancet2017Document17 pagesCAH Lancet2017telefontest118No ratings yet
- Fendo 11 596554 1Document17 pagesFendo 11 596554 1garrobosNo ratings yet
- Chapter 9 - Beta-Cell Neoplasia InsulinomaDocument28 pagesChapter 9 - Beta-Cell Neoplasia InsulinomaSteffi AraujoNo ratings yet
- Disorders of Pituitary Gland: Week 10Document13 pagesDisorders of Pituitary Gland: Week 10Irish Eunice FelixNo ratings yet
- In Cosmetology and TrichologyDocument72 pagesIn Cosmetology and TrichologySasi AttiliNo ratings yet
- 2009 AC 1 Monossomia Do 18qDocument6 pages2009 AC 1 Monossomia Do 18qedisonNo ratings yet
- Endocrine System g6 ReportingDocument122 pagesEndocrine System g6 ReportingEma Hibaya100% (1)
- PGD and PGS: Indications, Clinical Management, Results and Future PerspectivesDocument51 pagesPGD and PGS: Indications, Clinical Management, Results and Future Perspectiveslina ukago100% (1)
- Pituitary Adenoma FinalDocument44 pagesPituitary Adenoma FinalTuhinaRaj100% (1)
- Endo - TransesDocument8 pagesEndo - TransesMelanie ManuzonNo ratings yet
- A695 PDFDocument9 pagesA695 PDFFauzi NoviaNo ratings yet
- RollNo - 02 PTSM - II Teratogenicity & GenotoxicityDocument55 pagesRollNo - 02 PTSM - II Teratogenicity & GenotoxicityJayesh KadamNo ratings yet
- LEC 03 - Pituitary Tumours PDFDocument54 pagesLEC 03 - Pituitary Tumours PDFIoana CozmaNo ratings yet
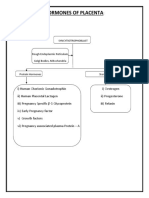
- Hormones of Placenta: SyncytiotrophoblastDocument3 pagesHormones of Placenta: SyncytiotrophoblastPabhat KumarNo ratings yet
- Disorders of Growth and DevelopmentDocument52 pagesDisorders of Growth and DevelopmentMaria Hudson100% (1)
- Obstetrics and GynecologyDocument64 pagesObstetrics and GynecologyLyk TiglaoNo ratings yet
- Nejmoa 066494Document11 pagesNejmoa 066494Alessandre AldiansyahNo ratings yet
- Gigantism and AcromegalyDocument5 pagesGigantism and AcromegalyAmoroso, Marian Corneth D.No ratings yet
- Congenital Adrenal HyperplasiaDocument17 pagesCongenital Adrenal HyperplasiaJeff CrocombeNo ratings yet
- Male Infertility PresentasiDocument23 pagesMale Infertility PresentasidewiNo ratings yet
- The Role of Targeted Therapy Ruxolitinib in The Management of PVcase SharingDocument30 pagesThe Role of Targeted Therapy Ruxolitinib in The Management of PVcase SharingMadjlusNo ratings yet
- AACE Clinical Case ReportsDocument5 pagesAACE Clinical Case ReportsSameerNo ratings yet
- LEC 03 - Pituitary TumoursDocument54 pagesLEC 03 - Pituitary TumoursIoana CozmaNo ratings yet
- PBL Week 1 Jumat EndocrineDocument29 pagesPBL Week 1 Jumat EndocrineHelena PadilahNo ratings yet
- A Model for Gene Therapy: Gene Replacement in the Treatment of Sickle Cell Anemia and ThalassemiaFrom EverandA Model for Gene Therapy: Gene Replacement in the Treatment of Sickle Cell Anemia and ThalassemiaNo ratings yet
- The Cardiovascular System: The Heart: Anatomy and PhysiologyDocument30 pagesThe Cardiovascular System: The Heart: Anatomy and PhysiologyZachary CohenNo ratings yet
- Atrial Fibrillation CASE STUDYDocument2 pagesAtrial Fibrillation CASE STUDYChristianneMikeNo ratings yet
- Effect of Corona Pandemic On Hospitality and Tourism IndustryDocument3 pagesEffect of Corona Pandemic On Hospitality and Tourism IndustryInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- AnesthesiaDocument10 pagesAnesthesiaMohamed EssamNo ratings yet
- Perbedaan Kadar Kolesterol Sebelum Dan Sesudah Dilakukan Brisk Walking Pada SiswaDocument8 pagesPerbedaan Kadar Kolesterol Sebelum Dan Sesudah Dilakukan Brisk Walking Pada SiswaEva RahmadaniNo ratings yet
- TracheostomyDocument29 pagesTracheostomyFemi AustinNo ratings yet
- Yersinia Pestis V Protein Epitopes Recognized by CD4 T CellsDocument8 pagesYersinia Pestis V Protein Epitopes Recognized by CD4 T CellsHerdi ArdianaNo ratings yet
- Early Complications of MIDocument11 pagesEarly Complications of MIjen262004No ratings yet
- Astm E2361 - 1 (En)Document5 pagesAstm E2361 - 1 (En)Sainath AmudaNo ratings yet
- Prelims NutridietDocument6 pagesPrelims Nutridietuma crespoNo ratings yet
- Armitage 2013 ReviewDocument17 pagesArmitage 2013 ReviewMarcoNo ratings yet
- What Is Osteopenia?: What Are Causes and Risk Factors For Osteopenia?Document2 pagesWhat Is Osteopenia?: What Are Causes and Risk Factors For Osteopenia?Jeremy EvansNo ratings yet
- Internal Medicine End of Rotation QuestionsDocument4 pagesInternal Medicine End of Rotation Questionskt_one100% (1)
- International Council of Ophthalmology's Ophthalmology Surgical Competency Assessment Rubric (ICO-OSCAR)Document8 pagesInternational Council of Ophthalmology's Ophthalmology Surgical Competency Assessment Rubric (ICO-OSCAR)Jose Antonio Fuentes VegaNo ratings yet
- Consciousness Traumatic Brain Injury: Eye Opening (E)Document3 pagesConsciousness Traumatic Brain Injury: Eye Opening (E)Nathalie kate petallarNo ratings yet
- Role of Albumin in Cirrhosis: From A Hospitalist's PerspectiveDocument8 pagesRole of Albumin in Cirrhosis: From A Hospitalist's PerspectivePutri PitalokaNo ratings yet
- Scaphoid Fracture: A Case ReportDocument5 pagesScaphoid Fracture: A Case Reportrapannika100% (2)
- EBOOK Modern Nutrition in Health and Disease Modern Nutrition in Health Disease Shils 11Th Edition Ebook PDF Download Full Chapter PDF KindleDocument62 pagesEBOOK Modern Nutrition in Health and Disease Modern Nutrition in Health Disease Shils 11Th Edition Ebook PDF Download Full Chapter PDF Kindlecarla.robbins451100% (45)
- A Comparison of Local and in The of Herpetic Disciform KeratitisDocument4 pagesA Comparison of Local and in The of Herpetic Disciform KeratitisAndi Raodah ImranNo ratings yet
- Seminar: Benjamin E Schreiber, Charlotte Agrup, Dorian O Haskard, Linda M LuxonDocument9 pagesSeminar: Benjamin E Schreiber, Charlotte Agrup, Dorian O Haskard, Linda M Luxonmaria_garaveNo ratings yet
- The Effect of WarDocument16 pagesThe Effect of Warapi-251665102No ratings yet
- Answer All The Following Short Essay Questions: (3 Questions)Document4 pagesAnswer All The Following Short Essay Questions: (3 Questions)Soad ShedeedNo ratings yet
- Pariyanan Jaruchinda Department of Otolaryngology Phramongkutklao HospitalDocument40 pagesPariyanan Jaruchinda Department of Otolaryngology Phramongkutklao HospitalWidi HadiNo ratings yet
- What Is The Digestive System?Document5 pagesWhat Is The Digestive System?SlaheddineNo ratings yet
- Nasogastric TubeDocument78 pagesNasogastric TubeQuia Benjch Uayan100% (1)
- Combined Science b8Document6 pagesCombined Science b8Jose ArcosNo ratings yet
- Sexual Dysfunction: By: Dr. Cecep Sugeng Kristanto SP KJ (K)Document30 pagesSexual Dysfunction: By: Dr. Cecep Sugeng Kristanto SP KJ (K)Rina WidarsihNo ratings yet