Download as pdf or txt
You might also like
- S-Scan Service Manual 141012700 Rev2 SW3.1A (Feb2014)Document165 pagesS-Scan Service Manual 141012700 Rev2 SW3.1A (Feb2014)wilton alves da silva100% (1)
- Instrumental Methods of Drug AnalysisFrom EverandInstrumental Methods of Drug AnalysisRating: 3 out of 5 stars3/5 (3)
- Multiple Chromatographic Fingerprinting and Its Application To The Quality Control of Herbal MedicinesDocument8 pagesMultiple Chromatographic Fingerprinting and Its Application To The Quality Control of Herbal MedicinesDanilo FlumignanNo ratings yet
- Anti-Tumor Activity of Annona Squamosa Seeds Extract Containing Annonaceous Acetogenin Compounds PDFDocument6 pagesAnti-Tumor Activity of Annona Squamosa Seeds Extract Containing Annonaceous Acetogenin Compounds PDFElizabeth Noel100% (1)
- Identification of The Phenolic Components of Chrysanthemum FlowerDocument8 pagesIdentification of The Phenolic Components of Chrysanthemum FlowerYou Jin LimNo ratings yet
- Food Chemistry: A B A A A ADocument7 pagesFood Chemistry: A B A A A AdickyNo ratings yet
- 21.VJST 19 57 (2) 155-161Document7 pages21.VJST 19 57 (2) 155-161Đào Ngọc Thuý VyNo ratings yet
- Anita Wagh-39-Vol.-10-Issue-6-June-2019-IJPSR-RA-11001 PDFDocument6 pagesAnita Wagh-39-Vol.-10-Issue-6-June-2019-IJPSR-RA-11001 PDFbutlesrNo ratings yet
- Anti-Cancer and Antioxidant Properties of Phenolics Isolated From Toona Sinensis A Juss Acetone Leaf ExtractDocument9 pagesAnti-Cancer and Antioxidant Properties of Phenolics Isolated From Toona Sinensis A Juss Acetone Leaf ExtractGrace Anastasia Ginting SinusingaNo ratings yet
- Galico AcidoDocument6 pagesGalico AcidoMaría Eugenia OlivaresNo ratings yet
- Article 02Document12 pagesArticle 02Sana AjmalNo ratings yet
- Trace Determination of Pharmaceuticals and Other Wastewater-Derived Micropollutants by Solid Phase Extraction and Gas Chromatography/mass SpectrometryDocument7 pagesTrace Determination of Pharmaceuticals and Other Wastewater-Derived Micropollutants by Solid Phase Extraction and Gas Chromatography/mass SpectrometryJosé SendimNo ratings yet
- Peel Citrus Reticulata - Foodchem.Document27 pagesPeel Citrus Reticulata - Foodchem.Aisyah MuthmainahNo ratings yet
- Journal: of Pharmaceutical ResearchDocument6 pagesJournal: of Pharmaceutical ResearchmeilaNo ratings yet
- Chemical Composition of Chrysanthemum Teas and Their Anti Inflammatory and Antioxidant PropertiesDocument9 pagesChemical Composition of Chrysanthemum Teas and Their Anti Inflammatory and Antioxidant PropertiesYou Jin LimNo ratings yet
- Rapid Screening and Identification of Antioxidants in Aqueous Ex - 2009 - Food CDocument7 pagesRapid Screening and Identification of Antioxidants in Aqueous Ex - 2009 - Food CTheerapat Chalerm-MuangNo ratings yet
- IJPhS 70 96Document4 pagesIJPhS 70 96MANUEL ALEJANDRO CHACON FUENTESNo ratings yet
- HPLC Determination of Bioactive Flavonoids in Hovenia Dulcis Fruit ExtractsDocument6 pagesHPLC Determination of Bioactive Flavonoids in Hovenia Dulcis Fruit ExtractsDewi ParamitaNo ratings yet
- Antioxidant and Cytotoxic Effects of Methanol Extracts of AmorphophallusDocument4 pagesAntioxidant and Cytotoxic Effects of Methanol Extracts of AmorphophallusDidar SadiqNo ratings yet
- Β - Sitosterol, Lupeol, Betulin, 2-Methyl Anthraquinones, Corchoroside-A And Fusidic Acid From Corchorusaestuan RootsDocument5 pagesΒ - Sitosterol, Lupeol, Betulin, 2-Methyl Anthraquinones, Corchoroside-A And Fusidic Acid From Corchorusaestuan Rootsmehedish09No ratings yet
- J.Pharm - Biomed.Anal. HPTLCandHPLC CalendulaOfficinalis 2014Document9 pagesJ.Pharm - Biomed.Anal. HPTLCandHPLC CalendulaOfficinalis 2014vikram kushwahaNo ratings yet
- Crystallization Org Process 2010 QuDocument7 pagesCrystallization Org Process 2010 QuMarcela TapiasNo ratings yet
- s00764 023 00230 7Document9 pagess00764 023 00230 7Artem KulikovNo ratings yet
- Analysis of Phytochemical Constituents and AntimicDocument6 pagesAnalysis of Phytochemical Constituents and AntimicRussel AloceljaNo ratings yet
- HPLC Ganoderic AcidDocument6 pagesHPLC Ganoderic AcidHu MihiNo ratings yet
- Cytotoxic Activity Screening of Some Indigenous THDocument5 pagesCytotoxic Activity Screening of Some Indigenous THAngelina KobanNo ratings yet
- Chemical Constituents of Corchorus Olitorius LDocument5 pagesChemical Constituents of Corchorus Olitorius LCyclopes BlackmoorNo ratings yet
- 2009 - Flavour Components and Antioxidant Properties of Several Cultivated MushroomsDocument7 pages2009 - Flavour Components and Antioxidant Properties of Several Cultivated MushroomsTrungTâmYaSaNo ratings yet
- Simultaneous determination of lupeol and β-sitosterol by high-performance thin-layer chromatographic method in Crataeva nurvala Buch-Ham. stem barkDocument6 pagesSimultaneous determination of lupeol and β-sitosterol by high-performance thin-layer chromatographic method in Crataeva nurvala Buch-Ham. stem barkArtem KulikovNo ratings yet
- CascaraDocument10 pagesCascaraVitor MiguesNo ratings yet
- NaprazniculDocument7 pagesNaprazniculIrina GurdisNo ratings yet
- 5c0a PDFDocument9 pages5c0a PDFnelisaNo ratings yet
- Thin-Layer Chromatographic Separation and Validated HPTLC MethodDocument8 pagesThin-Layer Chromatographic Separation and Validated HPTLC MethodAjay BhoyeNo ratings yet
- Four New C-Benzyl Flavonoids From The Fruit of Uvaria Cherrevensis.Document5 pagesFour New C-Benzyl Flavonoids From The Fruit of Uvaria Cherrevensis.shaniNo ratings yet
- địa liền inflammatory PDFDocument8 pagesđịa liền inflammatory PDFTâm PhanNo ratings yet
- Chiral Screening Approach of Atorvastatin Diastereomers by HPLC MethodDocument5 pagesChiral Screening Approach of Atorvastatin Diastereomers by HPLC MethodMediterr J Pharm Pharm SciNo ratings yet
- Phytochemical Screening by LC-MS Analysis of Flowers Of: Allamanda Neriifolia HookDocument9 pagesPhytochemical Screening by LC-MS Analysis of Flowers Of: Allamanda Neriifolia HookAi herlinaNo ratings yet
- 2023 - Journal Os Separation Science - Pesticide Residues in Herbs and Their Transfer For InfusionsDocument11 pages2023 - Journal Os Separation Science - Pesticide Residues in Herbs and Their Transfer For InfusionsLucas CaldeirãoNo ratings yet
- Food ChemistryDocument9 pagesFood ChemistryLuqman MileNo ratings yet
- Research Article: Development of Micellar HPLC-UV Method For Determination of Pharmaceuticals in Water SamplesDocument13 pagesResearch Article: Development of Micellar HPLC-UV Method For Determination of Pharmaceuticals in Water Samplesadolfo olmosNo ratings yet
- ChitosanDocument8 pagesChitosanmaria dulceNo ratings yet
- Phytochemical Analysis and Comprehensive Evaluation of Antimicrobial and Antioxidant Properties of 61 Medicinal Plant SpeciesDocument13 pagesPhytochemical Analysis and Comprehensive Evaluation of Antimicrobial and Antioxidant Properties of 61 Medicinal Plant SpeciesNichole Evita PascuaNo ratings yet
- 05 Intan Soraya Che Sulaiman - Paling FunctionDocument14 pages05 Intan Soraya Che Sulaiman - Paling FunctionIdham ZaharudieNo ratings yet
- Method Development and Validation of Daclatasvir in Bulk & Pharmaceutical Dosage Form by Uv-Visible SpectrophotometryDocument6 pagesMethod Development and Validation of Daclatasvir in Bulk & Pharmaceutical Dosage Form by Uv-Visible SpectrophotometryBaru Chandrasekhar RaoNo ratings yet
- Quantitative Analysis of Quercetin in Natural Sources by RPHPLCDocument4 pagesQuantitative Analysis of Quercetin in Natural Sources by RPHPLCjasp10agostoNo ratings yet
- Food Chemistry: Da-Wei Li, Ming Zhu, Yun-Dong Shao, Zhe Shen, Chen-Chen Weng, Wei-Dong YanDocument9 pagesFood Chemistry: Da-Wei Li, Ming Zhu, Yun-Dong Shao, Zhe Shen, Chen-Chen Weng, Wei-Dong YanTrần Hồ Hữu LuânNo ratings yet
- Amornrat san,+Journal+manager,+SMJ+v3+240-245+RattanaDocument6 pagesAmornrat san,+Journal+manager,+SMJ+v3+240-245+Rattananoviantyramadhani12No ratings yet
- GC-MS Analysis of Phytocomponents On Whole Plant Extract Adiantum Capillus-Veneris L. - A Potential Folklore Medicinal PlantDocument6 pagesGC-MS Analysis of Phytocomponents On Whole Plant Extract Adiantum Capillus-Veneris L. - A Potential Folklore Medicinal PlantRigotti BrNo ratings yet
- Thymol HPLC PDFDocument7 pagesThymol HPLC PDFdonigawantoNo ratings yet
- Accepted Manuscript: Food ChemistryDocument30 pagesAccepted Manuscript: Food ChemistryMisganaw AndualemNo ratings yet
- Articulo 06Document7 pagesArticulo 06iria.gonzalez.micoNo ratings yet
- Spectrophotometric Method For Polyphenols Analysis Pre Validation and Application On Plantago L SpeciesDocument6 pagesSpectrophotometric Method For Polyphenols Analysis Pre Validation and Application On Plantago L SpeciesVan CarlosNo ratings yet
- Artigo 2Document7 pagesArtigo 2ELISANGELA SILVANo ratings yet
- Jurnal Kpsel 62-64-66Document7 pagesJurnal Kpsel 62-64-66Hana NopiaNo ratings yet
- 10.1007@s00764 019 00004 0Document7 pages10.1007@s00764 019 00004 0Miy AichNo ratings yet
- Chemical Fingerprint Analysis and Quantitative Analysis of Rosa Rugosa by UPLC-DADDocument11 pagesChemical Fingerprint Analysis and Quantitative Analysis of Rosa Rugosa by UPLC-DADHuy BakNo ratings yet
- tmp4359 TMPDocument12 pagestmp4359 TMPFrontiersNo ratings yet
- D Jothiewari Et. Al.. (UV)Document5 pagesD Jothiewari Et. Al.. (UV)EVELYN SOLANHS ACERO RODRIGUEZNo ratings yet
- HPTLC Fingerprint Profiles and UPLCMS Identification of Potential Anti-Oxidant Fractions and Compounds From Ambrosia Martima and Ammi Majus L.Document10 pagesHPTLC Fingerprint Profiles and UPLCMS Identification of Potential Anti-Oxidant Fractions and Compounds From Ambrosia Martima and Ammi Majus L.Mona SalihNo ratings yet
- SimultaneousDocument5 pagesSimultaneousTrần ChâuNo ratings yet
- High Performance Liquid Chromatography Method ForDocument8 pagesHigh Performance Liquid Chromatography Method ForWalther YepesNo ratings yet
- The Rainmaker DesignDocument5 pagesThe Rainmaker DesignYến NhiNo ratings yet
- SpeakingDocument2 pagesSpeakingYến NhiNo ratings yet
- Ol902971w Si 002Document18 pagesOl902971w Si 002Yến NhiNo ratings yet
- Journal of Ethnopharmacology: Xidan Zhou, Liying Tang, Yilong Xu, Guohong Zhou, Zhuju WangDocument17 pagesJournal of Ethnopharmacology: Xidan Zhou, Liying Tang, Yilong Xu, Guohong Zhou, Zhuju WangYến NhiNo ratings yet
- Case Study Tourism New Zealand WebsiteDocument15 pagesCase Study Tourism New Zealand WebsiteYến NhiNo ratings yet
- 3b.boiler Treatment MethodsDocument76 pages3b.boiler Treatment Methodsalokbdas100% (1)
- FinitosDocument772 pagesFinitosVictor Jesus Rios100% (5)
- Analysing The Combination and Positioning of Bio Fibre and Bio FibreDocument6 pagesAnalysing The Combination and Positioning of Bio Fibre and Bio FibreAadarsh SharmaNo ratings yet
- Templates: Code Sharing (Genericity)Document9 pagesTemplates: Code Sharing (Genericity)killer crewmateNo ratings yet
- 8051MICROCONTROLLER BASED GAS AND FIRE ALARM SYSTEM+final RepDocument80 pages8051MICROCONTROLLER BASED GAS AND FIRE ALARM SYSTEM+final RepBhuwon Arjun83% (6)
- Manual Unity ProDocument726 pagesManual Unity ProMagno MonteiroNo ratings yet
- The Holy Grail Strategy FinderDocument35 pagesThe Holy Grail Strategy FinderFrancesco Nano100% (1)
- GIS Software IntroductionDocument28 pagesGIS Software IntroductionMian Waqar Ali ShahNo ratings yet
- Lieber 1961 Human Values and Science Art and MathematicsDocument74 pagesLieber 1961 Human Values and Science Art and MathematicsaNo ratings yet
- Tutorial 1 (Chapter 11a)Document2 pagesTutorial 1 (Chapter 11a)EcoliNo ratings yet
- Wiseco 16ST Engine 999 1071Document73 pagesWiseco 16ST Engine 999 1071Danno N0% (1)
- Let's Make A Decision..: Situation? APC - Alternatives, Possibilities and Choices 1. 2. 3. PrioritizeDocument6 pagesLet's Make A Decision..: Situation? APC - Alternatives, Possibilities and Choices 1. 2. 3. PrioritizekalaiNo ratings yet
- NSCV Part C-6a-Intact Stability RequirementsDocument111 pagesNSCV Part C-6a-Intact Stability RequirementsNgoc BangNo ratings yet
- Nutanix Collector User Guide v4 - 0Document48 pagesNutanix Collector User Guide v4 - 0Clement EbereNo ratings yet
- 12 Physics Notes Ch10 Wave OpticsDocument10 pages12 Physics Notes Ch10 Wave OpticsKOMAL NAVARIYANo ratings yet
- Chapter 5.9 - Sanitary SystemDocument29 pagesChapter 5.9 - Sanitary SystemHussen MohammedNo ratings yet
- Passive VoiceDocument2 pagesPassive VoiceDho FajNo ratings yet
- Driling ProcessDocument49 pagesDriling ProcessfmebirimNo ratings yet
- Methodology of The Canadian Labour Force SurveyDocument109 pagesMethodology of The Canadian Labour Force Survey42度鱼No ratings yet
- Model 1Document59 pagesModel 1Nut Name46No ratings yet
- Aa Microlab300 Cat 8Document52 pagesAa Microlab300 Cat 8DARWIN FERNANDO MONTOYA GONZALEZNo ratings yet
- SyllabusDocument10 pagesSyllabusroy rockNo ratings yet
- Manual de Perforadoras JacklegDocument34 pagesManual de Perforadoras JacklegFernando Pretell LozanoNo ratings yet
- Bending of PlatesDocument72 pagesBending of PlatesPrajeesh Raj100% (1)
- EigenFunctions, EigenValues & ExamplesDocument5 pagesEigenFunctions, EigenValues & ExamplesFajar Emman AsmatNo ratings yet
- WPS D1.3 RF 40Document8 pagesWPS D1.3 RF 40RodolfoNo ratings yet
- Atomic Structure and Interatomic BondingDocument12 pagesAtomic Structure and Interatomic BondingNicole Irene Dela PenaNo ratings yet
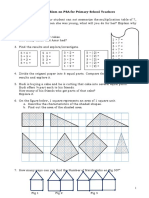
- Problem On PSA Primary SchoolDocument2 pagesProblem On PSA Primary SchoolMangleswary SalbarajaNo ratings yet