Download as pdf or txt
You might also like
- Short Lab Report Sheet Marcet Boiler 2021Document2 pagesShort Lab Report Sheet Marcet Boiler 2021DharmaalManieNo ratings yet
- 1 - Taylor, Cheng - 2010 - Economizer High Limit Controls and Why Enthalpy Economizers Don't WorkDocument17 pages1 - Taylor, Cheng - 2010 - Economizer High Limit Controls and Why Enthalpy Economizers Don't WorkpedroguaraldiNo ratings yet
- حل المسائل کتاب رفتار مکانیکی مواد نورمن دولینگ - ویرایش سومDocument7 pagesحل المسائل کتاب رفتار مکانیکی مواد نورمن دولینگ - ویرایش سومengineerNo ratings yet
- Vapor Liquid EquilibriaDocument22 pagesVapor Liquid Equilibriaanwarabdullah960No ratings yet
- Chapter 12Document38 pagesChapter 12Sudha CNo ratings yet
- CHE 631: Chemical Reaction Engineering: Animangsu GhatakDocument11 pagesCHE 631: Chemical Reaction Engineering: Animangsu GhatakPankaj Kumar SainiNo ratings yet
- Thermodynamic Property RelationsDocument37 pagesThermodynamic Property RelationsArunNo ratings yet
- DistillationDocument51 pagesDistillationsampathkumarNo ratings yet
- Chapter 7 Lecture Notes: DN N X DN N X DN N X DXDocument13 pagesChapter 7 Lecture Notes: DN N X DN N X DN N X DXNisa ShiningNo ratings yet
- Ch6 - MixturesDocument63 pagesCh6 - MixturesShaktivell Letchumanan100% (1)
- Solution NotesDocument17 pagesSolution NotesBabli KumariNo ratings yet
- Maxwell Relations 1Document39 pagesMaxwell Relations 1abdulqaderNo ratings yet
- 2008 Physical Chemistry 3Document52 pages2008 Physical Chemistry 3julianodesouzaNo ratings yet
- Glycol Dehydration of High-Acid Gas StreamsDocument10 pagesGlycol Dehydration of High-Acid Gas StreamsAndri SaputraNo ratings yet
- Behaviour of GasesDocument29 pagesBehaviour of GasesAli RazaNo ratings yet
- Chemistry 2402 - Thermodynamics: Lecture 13: Non Ideal Solutions and ActivityDocument13 pagesChemistry 2402 - Thermodynamics: Lecture 13: Non Ideal Solutions and ActivityZahurul IslamNo ratings yet
- Chapter 04 Physical TransformationsDocument40 pagesChapter 04 Physical TransformationsRuby DalyNo ratings yet
- CHE201 - Chapter 6 Dr. Nabeel Abo-Ghander NotesDocument39 pagesCHE201 - Chapter 6 Dr. Nabeel Abo-Ghander NotesMohammed AlshangitiNo ratings yet
- Solution NotesDocument110 pagesSolution NotesMomosNo ratings yet
- MLZ 325 Solution Thermodynamics Chapter 9 1 Raoults Henrys LawDocument27 pagesMLZ 325 Solution Thermodynamics Chapter 9 1 Raoults Henrys Lawerdem2tanjuNo ratings yet
- 5310 Mass Diffusion and Droplet EvaporationDocument8 pages5310 Mass Diffusion and Droplet EvaporationTushar PanchalNo ratings yet
- PT Chapter 6Document64 pagesPT Chapter 6shubhamNo ratings yet
- Gas Liquid ReactorsDocument20 pagesGas Liquid ReactorsjamNo ratings yet
- L7-Properties of Dry GasesDocument35 pagesL7-Properties of Dry GasesRakesh SinghNo ratings yet
- CHAPTER 5.1 GasDocument18 pagesCHAPTER 5.1 GasZARITH SOFHIA BINTI MD KHARODIN KM-PelajarNo ratings yet
- Chemical Equilibrium FDocument13 pagesChemical Equilibrium FRaju SinghNo ratings yet
- Thermodynamic PropertiesDocument20 pagesThermodynamic Propertiesmajd hussinNo ratings yet
- Development of Eos For Vapours & Gases: Models For Highly Bountiful Phase .. .Document28 pagesDevelopment of Eos For Vapours & Gases: Models For Highly Bountiful Phase .. .Muket AgmasNo ratings yet
- 10 Most Important Derivations With SolutionsDocument18 pages10 Most Important Derivations With SolutionsManish PattajoshiNo ratings yet
- 1basics of Process Calculations by Dr. Chetan M. PatelDocument10 pages1basics of Process Calculations by Dr. Chetan M. PatelYash JaiswalNo ratings yet
- Dr. J. VenkatesanDocument56 pagesDr. J. VenkatesanRajesh KumarNo ratings yet
- Properties of Pure Substance - 2Document49 pagesProperties of Pure Substance - 2Areyan HaqueNo ratings yet
- The Solubility of Organic Compounds in Supercritical CO2Document8 pagesThe Solubility of Organic Compounds in Supercritical CO2Davide Di ZioNo ratings yet
- Phase Diagrams of MixturesDocument24 pagesPhase Diagrams of MixturesmohammedNo ratings yet
- Chapter 2 فزيوكيميائيّةDocument22 pagesChapter 2 فزيوكيميائيّةrabushkhidemNo ratings yet
- HGE FormulaDocument35 pagesHGE FormulaMirajoy TardioNo ratings yet
- Thermodynamics 1: Volumetric Properties of Pure FluidsDocument24 pagesThermodynamics 1: Volumetric Properties of Pure FluidsHabib Faisal Yahya100% (1)
- Lecture 8 Phase EquilibriumDocument24 pagesLecture 8 Phase EquilibriumiB13eNo ratings yet
- 0705 2894 PDFDocument14 pages0705 2894 PDFHassanImranNo ratings yet
- Colligative Properties: Nathaniel P. DugosDocument32 pagesColligative Properties: Nathaniel P. DugossololexzibNo ratings yet
- States of Matter 14-10-2020 SynopsisDocument12 pagesStates of Matter 14-10-2020 SynopsisMvnmurthy ChikkalaNo ratings yet
- Physical Chemistry Second Attempt 2022Document97 pagesPhysical Chemistry Second Attempt 2022shreyaskumar467No ratings yet
- Chapter 3 Volumetric Properties of Pure Fluids PDFDocument8 pagesChapter 3 Volumetric Properties of Pure Fluids PDFNikko ManaleseNo ratings yet
- 3 Hydrocarbon Phase BehaviourDocument45 pages3 Hydrocarbon Phase BehaviourMD. ASIF ALL AZADNo ratings yet
- Heat Transfer and Hydraulic Resistance During Condensation of Steam in A Horizontal Tube and in A Bundle of TubesDocument13 pagesHeat Transfer and Hydraulic Resistance During Condensation of Steam in A Horizontal Tube and in A Bundle of TubesBhsr Karthik VarmaNo ratings yet
- Minimum Learning Material CBSE ChemistryDocument50 pagesMinimum Learning Material CBSE ChemistryAntur RakhaNo ratings yet
- #1 Dissociation of Propionic Acid Dimer Sept-7-2021Document14 pages#1 Dissociation of Propionic Acid Dimer Sept-7-2021Yun-Ru, Rose ChenNo ratings yet
- Gas Absorption CompleteDocument18 pagesGas Absorption CompleteRanbir NarainNo ratings yet
- Face Your Challenge, Be Smart: JULY 20, 2013 Moscow, RussiaDocument38 pagesFace Your Challenge, Be Smart: JULY 20, 2013 Moscow, RussiaPhạm HưngNo ratings yet
- Physics 23 Fall 1993 Lab 2 - Adiabatic Processes: PV NRTDocument13 pagesPhysics 23 Fall 1993 Lab 2 - Adiabatic Processes: PV NRTvipul ch v v n s sNo ratings yet
- Experiment 6 Vapor Pressure of Pure LiquidsDocument8 pagesExperiment 6 Vapor Pressure of Pure LiquidsHaNo ratings yet
- Equations of StateDocument33 pagesEquations of StateDevika BharathanNo ratings yet
- MT-29.03.17 - Solutions and Phase DiagramsDocument96 pagesMT-29.03.17 - Solutions and Phase DiagramsSidharth JessyNo ratings yet
- UC Berkeley Previously Published WorksDocument12 pagesUC Berkeley Previously Published WorksMauricio Fabian Duque DazaNo ratings yet
- Chapter 4 - Gas AbsorptionDocument95 pagesChapter 4 - Gas AbsorptionBoyHaha100% (7)
- Chemical Equilibrium 1-CombinedDocument27 pagesChemical Equilibrium 1-CombinedCharlotte HooperNo ratings yet
- Solution Thermodynamics Theory-Ch 11Document50 pagesSolution Thermodynamics Theory-Ch 11Donni Azhar100% (2)
- ME2121 Thermodynamics: Gas-Vapour MixturesDocument5 pagesME2121 Thermodynamics: Gas-Vapour MixturesDesiree LinNo ratings yet
- Phase EquilibriaDocument21 pagesPhase EquilibriaGianna Cloe100% (1)
- SCH 103 NotesDocument50 pagesSCH 103 NotesJacqueseNo ratings yet
- Chemistry: SolutionDocument68 pagesChemistry: SolutionSatyajit RoutNo ratings yet
- Chapter 6Document44 pagesChapter 6Annerlynn SolanoNo ratings yet
- Working Guide to Vapor-Liquid Phase Equilibria CalculationsFrom EverandWorking Guide to Vapor-Liquid Phase Equilibria CalculationsRating: 5 out of 5 stars5/5 (1)
- حل المسائل مبانی ترمودینامیک مهندسی مایکل موران ویرایش هشتمDocument30 pagesحل المسائل مبانی ترمودینامیک مهندسی مایکل موران ویرایش هشتمengineerNo ratings yet
- ME 4214 Mechanical Behavior of Materials (Elective) : Catalog DescriptionDocument2 pagesME 4214 Mechanical Behavior of Materials (Elective) : Catalog DescriptionengineerNo ratings yet
- Mechanical Material2Document15 pagesMechanical Material2engineerNo ratings yet
- Software and Algorithms: - Programs Are Termed Software Because They Do Not Exist in A Physical, Tangible FormDocument27 pagesSoftware and Algorithms: - Programs Are Termed Software Because They Do Not Exist in A Physical, Tangible FormengineerNo ratings yet
- Electron Probe Microanalysis (EPMA) Measurement of Thin-Film Thickness in The Nanometre RangeDocument5 pagesElectron Probe Microanalysis (EPMA) Measurement of Thin-Film Thickness in The Nanometre RangeengineerNo ratings yet
- Problem Set 2Document2 pagesProblem Set 2engineerNo ratings yet
- Problem Set 3Document1 pageProblem Set 3engineerNo ratings yet
- 3.003 Principles of Engineering Practice: Light Materials AR Coatings Solar CellsDocument14 pages3.003 Principles of Engineering Practice: Light Materials AR Coatings Solar CellsengineerNo ratings yet
- Massachusetts Institute of TechnologyDocument2 pagesMassachusetts Institute of TechnologyengineerNo ratings yet
- The Linux ShellDocument20 pagesThe Linux ShellengineerNo ratings yet
- 3.032 Mechanical Behavior of Materials: Fall 2007Document3 pages3.032 Mechanical Behavior of Materials: Fall 2007engineerNo ratings yet
- 3.003 Principles of Engineering Practice: Light Materials AR Coatings Solar CellsDocument14 pages3.003 Principles of Engineering Practice: Light Materials AR Coatings Solar CellsengineerNo ratings yet
- Massachusetts Institute of TechnologyDocument1 pageMassachusetts Institute of TechnologyengineerNo ratings yet
- Construction MaterialsDocument3 pagesConstruction MaterialsengineerNo ratings yet
- Solutions To The Diffusion EquationDocument18 pagesSolutions To The Diffusion EquationengineerNo ratings yet
- ThermodynamicsDocument137 pagesThermodynamicsengineerNo ratings yet
- Bonnie NewmanDocument1 pageBonnie NewmanengineerNo ratings yet
- Tem PDFDocument90 pagesTem PDFengineerNo ratings yet
- Acid-Base EquilibriaDocument31 pagesAcid-Base EquilibriaKim Fan100% (1)
- Formative WorksheetDocument10 pagesFormative WorksheetMEGHNA BAGCHINo ratings yet
- Chemistry The Science in Context Volume I and II 5th Edition Gilbert Test BankDocument31 pagesChemistry The Science in Context Volume I and II 5th Edition Gilbert Test Bankjenniferrichardsonjrwfpzsdim100% (31)
- Pka BasesDocument1 pagePka BasesCarlos ArzaluzNo ratings yet
- Foundation Level Infrared Training Notes 12-Aug-2010Document92 pagesFoundation Level Infrared Training Notes 12-Aug-2010Sireesh AdimadhyamNo ratings yet
- Use of Heat and Mass Transfer Data Books, Steam Tables Are PermittedDocument4 pagesUse of Heat and Mass Transfer Data Books, Steam Tables Are Permitted3rajaNo ratings yet
- Tutorial 1Document2 pagesTutorial 1Aswath PNo ratings yet
- Thermal Properties of Pure Aluminium With Respect To TemperatureDocument4 pagesThermal Properties of Pure Aluminium With Respect To Temperatureahmedameer20189No ratings yet
- Chapter 3 - Examples and Exercises (Part II) NewDocument7 pagesChapter 3 - Examples and Exercises (Part II) NewtemesgenNo ratings yet
- Gen-Chem 2 - Worksheet-New-Template - Quarter 4-Week-2-Days-1-to-4Document7 pagesGen-Chem 2 - Worksheet-New-Template - Quarter 4-Week-2-Days-1-to-4CJ RhodesNo ratings yet
- Carnot CycleDocument26 pagesCarnot CycleNafisa AnikaNo ratings yet
- HT 1Document33 pagesHT 1aymaNo ratings yet
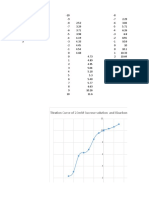
- Titration Curve of 20mM Sucrose Solution and Bicarbonate Buffer (PH of 9.8)Document6 pagesTitration Curve of 20mM Sucrose Solution and Bicarbonate Buffer (PH of 9.8)Hieu PhamNo ratings yet
- Stirred Tank ReactorsDocument38 pagesStirred Tank ReactorsJimNo ratings yet
- Thermochemistry (General Chem)Document50 pagesThermochemistry (General Chem)coppernitrateNo ratings yet
- HVAC ProductsDocument10 pagesHVAC ProductsMihai ConstantinescuNo ratings yet
- RPH Science ExperimentDocument5 pagesRPH Science ExperimentNorzilah MazaharNo ratings yet
- All 10Document11 pagesAll 10YacelinNo ratings yet
- Gibbs Free EnergyDocument12 pagesGibbs Free EnergyAyesha GulzarNo ratings yet
- Carta Sicrometrica A Nivel Del MarDocument1 pageCarta Sicrometrica A Nivel Del MarEstebanCórdobaNo ratings yet
- Question Bank of HeatDocument13 pagesQuestion Bank of HeatNaushaba Rangoonwala100% (1)
- Unit Conversion TablesDocument3 pagesUnit Conversion TablesMarin NicolaeNo ratings yet
- Thermo Activity 1,2,3Document3 pagesThermo Activity 1,2,3Karl Christian FajardoNo ratings yet
- Solcap6 PDFDocument70 pagesSolcap6 PDFLuiz Felipe Correa CardenasNo ratings yet
- Heat Bridges Catalog en 1205Document9 pagesHeat Bridges Catalog en 1205Душан ПузовићNo ratings yet
- Unit 4 - Equilibria Inckuding Acid Base QuestionsDocument134 pagesUnit 4 - Equilibria Inckuding Acid Base Questionsareyouthere92100% (1)
- GAPL-HVAC People UKDocument20 pagesGAPL-HVAC People UKrandeep ravishNo ratings yet
- SCI 104 Lecture 3 ThermochemistryDocument50 pagesSCI 104 Lecture 3 ThermochemistryYana100% (1)