
Banegas Luna2018
Banegas Luna2018
You might also like
- Summer Training Report On Python Cum Computer VisionDocument28 pagesSummer Training Report On Python Cum Computer Visionmd.sazid ansari100% (2)
- P6 Training AberdeenDocument3 pagesP6 Training AberdeenNnamdiNo ratings yet
- Virtual ScreeningDocument4 pagesVirtual ScreeningVinish ChoudaryNo ratings yet
- Molecular Descriptors and Methods For Ligand Based Virtual High Throughput Screening in Drug DiscoveryDocument13 pagesMolecular Descriptors and Methods For Ligand Based Virtual High Throughput Screening in Drug DiscoveryHeneampong IsaacNo ratings yet
- Target-Oriented Generic Fingerprint-Based Molecular RepresentationDocument14 pagesTarget-Oriented Generic Fingerprint-Based Molecular RepresentationCS & ITNo ratings yet
- J Swevo 2018 10 004Document40 pagesJ Swevo 2018 10 004Freddy Garcia FuentesNo ratings yet
- Large Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug DiscoveryDocument16 pagesLarge Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug DiscoverydashanunravanNo ratings yet
- 2017 Role of Computational Efficiency Indices and Pose Clustering in EffectiveDocument14 pages2017 Role of Computational Efficiency Indices and Pose Clustering in EffectiveFuckNo ratings yet
- Novel Hybrid Ultrafast Shape Descriptor Method For Use in Virtual ScreeningDocument10 pagesNovel Hybrid Ultrafast Shape Descriptor Method For Use in Virtual Screeningsatish kumarNo ratings yet
- Fulltext PDFDocument9 pagesFulltext PDFPace RaditNo ratings yet
- 1 s2.0 S0165993620303861 MainDocument21 pages1 s2.0 S0165993620303861 MainMuhammad Koksh Sdiq HussinNo ratings yet
- LikeDocument12 pagesLikespyrosgay33No ratings yet
- Bbad 014Document15 pagesBbad 014kanthaNo ratings yet
- Dock Around The Clock - Current Status of Small MoleculeDocument11 pagesDock Around The Clock - Current Status of Small Moleculetaoufik akabliNo ratings yet
- BasedpairedDocument34 pagesBasedpairedRicardo RomeroNo ratings yet
- OHagan-Kell2017 Article AnalysisOfDrugEndogenousHumanM PDFDocument17 pagesOHagan-Kell2017 Article AnalysisOfDrugEndogenousHumanM PDFYOUTEACHNo ratings yet
- Electronics 11 01125 v2Document31 pagesElectronics 11 01125 v2Shaniba HaneefaNo ratings yet
- MetaboliticsDB IEEE TCBB-2Document13 pagesMetaboliticsDB IEEE TCBB-2Hayri KulNo ratings yet
- Descritores e Seus Métodos de Seleção Na Análise QSAR: Paradigma para o Design de MedicamentosDocument12 pagesDescritores e Seus Métodos de Seleção Na Análise QSAR: Paradigma para o Design de MedicamentosLucas PalmeiraNo ratings yet
- Basic Overview of ChemoinformaticsDocument11 pagesBasic Overview of ChemoinformaticskinjalpcNo ratings yet
- Literature Review of Water Supply ProjectDocument6 pagesLiterature Review of Water Supply Projectdoawpfcnd100% (1)
- Beware of DockingDocument18 pagesBeware of DockingLeonardo RanderNo ratings yet
- Paper6 - Bionoinformatics NosqlDocument5 pagesPaper6 - Bionoinformatics NosqlmarcoNo ratings yet
- MolDivers200913 277285Document10 pagesMolDivers200913 277285RadioLaSounnaNo ratings yet
- Linking Clinical DataDocument6 pagesLinking Clinical DataNidhi GuptaNo ratings yet
- Processes 09 00950 v2Document16 pagesProcesses 09 00950 v2IITRANANDJADHAVNo ratings yet
- LimGen Ecml PKDDDocument19 pagesLimGen Ecml PKDDskannanauNo ratings yet
- CRAFTED - An Exploratory Database of Simulated Adsorption Isotherms of Metal-Organic FrameworksDocument18 pagesCRAFTED - An Exploratory Database of Simulated Adsorption Isotherms of Metal-Organic FrameworksZijun DengNo ratings yet
- Recent Advances in Computer-Aided Drug DesignDocument13 pagesRecent Advances in Computer-Aided Drug DesignSubhadip DasNo ratings yet
- ApplicationDocument18 pagesApplicationTiên PhạmNo ratings yet
- Ann 3Document8 pagesAnn 3Saul Antonio Montoya SerranoNo ratings yet
- A Self-Attention Based Message Passing Neural NetwDocument10 pagesA Self-Attention Based Message Passing Neural NetwDuc ThuyNo ratings yet
- Converting Relational Databases Into Object-Relational DatabasesDocument18 pagesConverting Relational Databases Into Object-Relational DatabasesLCO17 369 Mayank BimbraNo ratings yet
- Intro To Chemical DatabaseDocument5 pagesIntro To Chemical DatabaseTejinder SinghNo ratings yet
- ChemMine Tools An Online Service For AnaDocument6 pagesChemMine Tools An Online Service For Anayur fanNo ratings yet
- Paper 4Document12 pagesPaper 4Ahmed aboraiaNo ratings yet
- Artificial Intelligence in Chemistry and Drug DesignDocument7 pagesArtificial Intelligence in Chemistry and Drug DesignbiapNo ratings yet
- Using Metaboanalyst 5.0 For LC - Hrms Spectra Processing, Multi-Omics Integration and Covariate Adjustment of Global Metabolomics DataDocument29 pagesUsing Metaboanalyst 5.0 For LC - Hrms Spectra Processing, Multi-Omics Integration and Covariate Adjustment of Global Metabolomics DataIqbal PrawiraNo ratings yet
- CompimaDocument26 pagesCompimajeremytoh89No ratings yet
- A Review of Protein-Small Molecule Docking MethodsDocument16 pagesA Review of Protein-Small Molecule Docking Methodsnareshph28No ratings yet
- Fulltext Ajbe v1 Id1017Document3 pagesFulltext Ajbe v1 Id1017Ahammed Abu DilNo ratings yet
- Practical Guide To Large Scale DockingDocument36 pagesPractical Guide To Large Scale DockingVishnu KirthiNo ratings yet
- 26 Augmented Hill-Climb Increases Reinforcement Learning Efficiency For Language-Based de Novo Molecule Generation.Document22 pages26 Augmented Hill-Climb Increases Reinforcement Learning Efficiency For Language-Based de Novo Molecule Generation.felicitywxxNo ratings yet
- Statistical and Analytical Comparison of Multi-Criteria Decision-Making Techniques Under Fuzzy EnvironmentDocument26 pagesStatistical and Analytical Comparison of Multi-Criteria Decision-Making Techniques Under Fuzzy EnvironmentAdnan FauziRachmanNo ratings yet
- DatabaseDocument10 pagesDatabaseDAYANGKU SYAIDATUL FARHANA BINTI AWANGKU ABDUL RAZAK BF21110191No ratings yet
- Molecular Docking PaperDocument24 pagesMolecular Docking PaperChristian CastroNo ratings yet
- Service Identification Methods A Systematic Literature ReviewDocument8 pagesService Identification Methods A Systematic Literature Reviewc5p8vze7No ratings yet
- Molecules: Effect of Dataset Size and Train/Test Split Ratios in QSAR/QSPR Multiclass ClassificationDocument16 pagesMolecules: Effect of Dataset Size and Train/Test Split Ratios in QSAR/QSPR Multiclass ClassificationFantaNo ratings yet
- Molecular Docking: in Computer Aided Drug DesignDocument26 pagesMolecular Docking: in Computer Aided Drug DesignGravelandNo ratings yet
- 1 s2.0 S0165993623001103 MainDocument12 pages1 s2.0 S0165993623001103 MainMuhammad Koksh Sdiq HussinNo ratings yet
- Ontologies in Software Testing A Systematic Literature ReviewDocument8 pagesOntologies in Software Testing A Systematic Literature ReviewafmabkbhckmajgNo ratings yet
- Molecular Docking TheoryDocument21 pagesMolecular Docking Theoryolivia6669No ratings yet
- Collision Cross Section (CCS) As A Complementary Parameter To Characterize Human and Veterinary DrugsDocument12 pagesCollision Cross Section (CCS) As A Complementary Parameter To Characterize Human and Veterinary DrugsSoraya OchsNo ratings yet
- Muegge 2010Document46 pagesMuegge 2010Santi SuronoNo ratings yet
- Dock EcrDocument10 pagesDock Ecryin chanNo ratings yet
- NOREVA - Normalization and Evaluation of MS-based Metabolomics DataDocument9 pagesNOREVA - Normalization and Evaluation of MS-based Metabolomics Dataciborg1978No ratings yet
- 2014 Biomed Res Int 348725Document13 pages2014 Biomed Res Int 348725mbrylinskiNo ratings yet
- 1 s2.0 S0010465522001175 MainDocument20 pages1 s2.0 S0010465522001175 MainI Putu Adi PratamaNo ratings yet
- DBARKHICheminf7 20Document14 pagesDBARKHICheminf7 20Risnawa Puji AstutiNo ratings yet
- A Benchmark Driven Guide To Binding SiteDocument50 pagesA Benchmark Driven Guide To Binding SiteAlexNo ratings yet
- Life Cycle Inventory Analysis: Methods and DataFrom EverandLife Cycle Inventory Analysis: Methods and DataAndreas CirothNo ratings yet
- Portfolio - Food ArticlesDocument16 pagesPortfolio - Food ArticlesDONI DERMAWANNo ratings yet
- Affinity Chromatography: Principles and Applications: Sameh Magdeldin and Annette MoserDocument26 pagesAffinity Chromatography: Principles and Applications: Sameh Magdeldin and Annette MoserDONI DERMAWANNo ratings yet
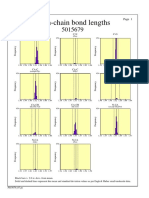
- Main-Chain Bond Lengths: ProcheckDocument1 pageMain-Chain Bond Lengths: ProcheckDONI DERMAWANNo ratings yet
- Tel: 1-631-626-9181 Fax: 1-631-614-7828Document11 pagesTel: 1-631-626-9181 Fax: 1-631-614-7828DONI DERMAWANNo ratings yet
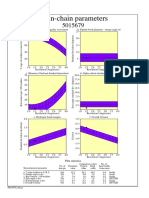
- Main-Chain Parameters: Plot StatisticsDocument1 pageMain-Chain Parameters: Plot StatisticsDONI DERMAWANNo ratings yet
- 2019-2020 Program Guide PDFDocument142 pages2019-2020 Program Guide PDFAndhy Arya EkaputraNo ratings yet
- Biomaterials Science: Volume 2 Number 4 April 2014 Pages 429-592Document9 pagesBiomaterials Science: Volume 2 Number 4 April 2014 Pages 429-592DONI DERMAWANNo ratings yet
- System PropertyDocument3 pagesSystem Propertymoveena abdullahNo ratings yet
- Computers For DummiesDocument14 pagesComputers For Dummiesapi-256251346No ratings yet
- Janak Raikhola CVDocument3 pagesJanak Raikhola CVsaroj khanalNo ratings yet
- Course Outline PF 2021Document4 pagesCourse Outline PF 2021Kabeer UddinNo ratings yet
- Queyquep MIDTERM PRACTICESET8Document10 pagesQueyquep MIDTERM PRACTICESET8annikaNo ratings yet
- 1011202222101PM-Class 5 Revision Worksheet For First Term Examination - MathsDocument7 pages1011202222101PM-Class 5 Revision Worksheet For First Term Examination - Mathsak dNo ratings yet
- Chapter 1: Introduction (PART 1) : Computer Networking: A Top Down Approach 6th Edition Jim Kurose, Keith RossDocument15 pagesChapter 1: Introduction (PART 1) : Computer Networking: A Top Down Approach 6th Edition Jim Kurose, Keith RosssylinxNo ratings yet
- SE25 49 DAW Setup GuideDocument5 pagesSE25 49 DAW Setup GuideDiego Ignacio Garcia JuradoNo ratings yet
- Libro Criollo SaxoDocument150 pagesLibro Criollo Saxohermogenes palominoNo ratings yet
- What Is The Smart Grid? Definitions, Perspectives, and Ultimate GoalsDocument6 pagesWhat Is The Smart Grid? Definitions, Perspectives, and Ultimate GoalsAhsan TariqNo ratings yet
- Best-Network-Security ToolsDocument7 pagesBest-Network-Security Toolsesa abrenNo ratings yet
- Cyclone V Device Handbook Volume 3 Hard Processor System Technical Reference Manual PDFDocument670 pagesCyclone V Device Handbook Volume 3 Hard Processor System Technical Reference Manual PDFMaximiliano NietoNo ratings yet
- MPLS L3VPN With Sub-Interface and VRF-Lite Between PE-CE - GNS3Document11 pagesMPLS L3VPN With Sub-Interface and VRF-Lite Between PE-CE - GNS3Camps PutraNo ratings yet
- Amjad Survey PaperDocument22 pagesAmjad Survey PaperDr-Mohammed RihanNo ratings yet
- Mathmatics 1ggggggggg PDFDocument753 pagesMathmatics 1ggggggggg PDFZahid Ali Mughal100% (2)
- ReadmeDocument2 pagesReadmematheus BarbosaNo ratings yet
- ISO8583 Message Converter OnlineDocument4 pagesISO8583 Message Converter Onlinesajad salehiNo ratings yet
- Computer Shopper August 2005 PDF eBook-iNTENSiTYDocument160 pagesComputer Shopper August 2005 PDF eBook-iNTENSiTYvoodoower100% (3)
- JNTUA-B.Tech.4-1 CSE-R15-SYLLABUSDocument36 pagesJNTUA-B.Tech.4-1 CSE-R15-SYLLABUSDr William AlbertNo ratings yet
- University PUNJAB (Gujranwala Campus) : Department Information Technology Academic Transcript (Information Technology)Document2 pagesUniversity PUNJAB (Gujranwala Campus) : Department Information Technology Academic Transcript (Information Technology)HAFIZ SHAHEER UL ISLAMNo ratings yet
- Backhaul RequirementsDocument25 pagesBackhaul RequirementsChetan BhatNo ratings yet
- Lecture1113 16473 AnimationDocument11 pagesLecture1113 16473 Animationashokgehlot2100No ratings yet
- Madda Walabu University: Collage of Computing Department of Information SystemDocument22 pagesMadda Walabu University: Collage of Computing Department of Information SystemLenchoNo ratings yet
- Ethernet Cable: Color Code Standards: Basic TheoryDocument3 pagesEthernet Cable: Color Code Standards: Basic TheoryPlacid ItyNo ratings yet
- IP Com 12Document23 pagesIP Com 12Tio_louis32No ratings yet
- TEE MTCSC292 Soft Computing LabDocument2 pagesTEE MTCSC292 Soft Computing LabEKNo ratings yet
- Item ConversionDocument7 pagesItem ConversionAnu RaadhaNo ratings yet
- Dr. Öğr. Üyesi Selçuk KİZİR 1Document71 pagesDr. Öğr. Üyesi Selçuk KİZİR 1DofilomNo ratings yet



























































































Download as pdf or txt
You might also like
- Summer Training Report On Python Cum Computer VisionDocument28 pagesSummer Training Report On Python Cum Computer Visionmd.sazid ansari100% (2)
- P6 Training AberdeenDocument3 pagesP6 Training AberdeenNnamdiNo ratings yet
- Virtual ScreeningDocument4 pagesVirtual ScreeningVinish ChoudaryNo ratings yet
- Molecular Descriptors and Methods For Ligand Based Virtual High Throughput Screening in Drug DiscoveryDocument13 pagesMolecular Descriptors and Methods For Ligand Based Virtual High Throughput Screening in Drug DiscoveryHeneampong IsaacNo ratings yet
- Target-Oriented Generic Fingerprint-Based Molecular RepresentationDocument14 pagesTarget-Oriented Generic Fingerprint-Based Molecular RepresentationCS & ITNo ratings yet
- J Swevo 2018 10 004Document40 pagesJ Swevo 2018 10 004Freddy Garcia FuentesNo ratings yet
- Large Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug DiscoveryDocument16 pagesLarge Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug DiscoverydashanunravanNo ratings yet
- 2017 Role of Computational Efficiency Indices and Pose Clustering in EffectiveDocument14 pages2017 Role of Computational Efficiency Indices and Pose Clustering in EffectiveFuckNo ratings yet
- Novel Hybrid Ultrafast Shape Descriptor Method For Use in Virtual ScreeningDocument10 pagesNovel Hybrid Ultrafast Shape Descriptor Method For Use in Virtual Screeningsatish kumarNo ratings yet
- Fulltext PDFDocument9 pagesFulltext PDFPace RaditNo ratings yet
- 1 s2.0 S0165993620303861 MainDocument21 pages1 s2.0 S0165993620303861 MainMuhammad Koksh Sdiq HussinNo ratings yet
- LikeDocument12 pagesLikespyrosgay33No ratings yet
- Bbad 014Document15 pagesBbad 014kanthaNo ratings yet
- Dock Around The Clock - Current Status of Small MoleculeDocument11 pagesDock Around The Clock - Current Status of Small Moleculetaoufik akabliNo ratings yet
- BasedpairedDocument34 pagesBasedpairedRicardo RomeroNo ratings yet
- OHagan-Kell2017 Article AnalysisOfDrugEndogenousHumanM PDFDocument17 pagesOHagan-Kell2017 Article AnalysisOfDrugEndogenousHumanM PDFYOUTEACHNo ratings yet
- Electronics 11 01125 v2Document31 pagesElectronics 11 01125 v2Shaniba HaneefaNo ratings yet
- MetaboliticsDB IEEE TCBB-2Document13 pagesMetaboliticsDB IEEE TCBB-2Hayri KulNo ratings yet
- Descritores e Seus Métodos de Seleção Na Análise QSAR: Paradigma para o Design de MedicamentosDocument12 pagesDescritores e Seus Métodos de Seleção Na Análise QSAR: Paradigma para o Design de MedicamentosLucas PalmeiraNo ratings yet
- Basic Overview of ChemoinformaticsDocument11 pagesBasic Overview of ChemoinformaticskinjalpcNo ratings yet
- Literature Review of Water Supply ProjectDocument6 pagesLiterature Review of Water Supply Projectdoawpfcnd100% (1)
- Beware of DockingDocument18 pagesBeware of DockingLeonardo RanderNo ratings yet
- Paper6 - Bionoinformatics NosqlDocument5 pagesPaper6 - Bionoinformatics NosqlmarcoNo ratings yet
- MolDivers200913 277285Document10 pagesMolDivers200913 277285RadioLaSounnaNo ratings yet
- Linking Clinical DataDocument6 pagesLinking Clinical DataNidhi GuptaNo ratings yet
- Processes 09 00950 v2Document16 pagesProcesses 09 00950 v2IITRANANDJADHAVNo ratings yet
- LimGen Ecml PKDDDocument19 pagesLimGen Ecml PKDDskannanauNo ratings yet
- CRAFTED - An Exploratory Database of Simulated Adsorption Isotherms of Metal-Organic FrameworksDocument18 pagesCRAFTED - An Exploratory Database of Simulated Adsorption Isotherms of Metal-Organic FrameworksZijun DengNo ratings yet
- Recent Advances in Computer-Aided Drug DesignDocument13 pagesRecent Advances in Computer-Aided Drug DesignSubhadip DasNo ratings yet
- ApplicationDocument18 pagesApplicationTiên PhạmNo ratings yet
- Ann 3Document8 pagesAnn 3Saul Antonio Montoya SerranoNo ratings yet
- A Self-Attention Based Message Passing Neural NetwDocument10 pagesA Self-Attention Based Message Passing Neural NetwDuc ThuyNo ratings yet
- Converting Relational Databases Into Object-Relational DatabasesDocument18 pagesConverting Relational Databases Into Object-Relational DatabasesLCO17 369 Mayank BimbraNo ratings yet
- Intro To Chemical DatabaseDocument5 pagesIntro To Chemical DatabaseTejinder SinghNo ratings yet
- ChemMine Tools An Online Service For AnaDocument6 pagesChemMine Tools An Online Service For Anayur fanNo ratings yet
- Paper 4Document12 pagesPaper 4Ahmed aboraiaNo ratings yet
- Artificial Intelligence in Chemistry and Drug DesignDocument7 pagesArtificial Intelligence in Chemistry and Drug DesignbiapNo ratings yet
- Using Metaboanalyst 5.0 For LC - Hrms Spectra Processing, Multi-Omics Integration and Covariate Adjustment of Global Metabolomics DataDocument29 pagesUsing Metaboanalyst 5.0 For LC - Hrms Spectra Processing, Multi-Omics Integration and Covariate Adjustment of Global Metabolomics DataIqbal PrawiraNo ratings yet
- CompimaDocument26 pagesCompimajeremytoh89No ratings yet
- A Review of Protein-Small Molecule Docking MethodsDocument16 pagesA Review of Protein-Small Molecule Docking Methodsnareshph28No ratings yet
- Fulltext Ajbe v1 Id1017Document3 pagesFulltext Ajbe v1 Id1017Ahammed Abu DilNo ratings yet
- Practical Guide To Large Scale DockingDocument36 pagesPractical Guide To Large Scale DockingVishnu KirthiNo ratings yet
- 26 Augmented Hill-Climb Increases Reinforcement Learning Efficiency For Language-Based de Novo Molecule Generation.Document22 pages26 Augmented Hill-Climb Increases Reinforcement Learning Efficiency For Language-Based de Novo Molecule Generation.felicitywxxNo ratings yet
- Statistical and Analytical Comparison of Multi-Criteria Decision-Making Techniques Under Fuzzy EnvironmentDocument26 pagesStatistical and Analytical Comparison of Multi-Criteria Decision-Making Techniques Under Fuzzy EnvironmentAdnan FauziRachmanNo ratings yet
- DatabaseDocument10 pagesDatabaseDAYANGKU SYAIDATUL FARHANA BINTI AWANGKU ABDUL RAZAK BF21110191No ratings yet
- Molecular Docking PaperDocument24 pagesMolecular Docking PaperChristian CastroNo ratings yet
- Service Identification Methods A Systematic Literature ReviewDocument8 pagesService Identification Methods A Systematic Literature Reviewc5p8vze7No ratings yet
- Molecules: Effect of Dataset Size and Train/Test Split Ratios in QSAR/QSPR Multiclass ClassificationDocument16 pagesMolecules: Effect of Dataset Size and Train/Test Split Ratios in QSAR/QSPR Multiclass ClassificationFantaNo ratings yet
- Molecular Docking: in Computer Aided Drug DesignDocument26 pagesMolecular Docking: in Computer Aided Drug DesignGravelandNo ratings yet
- 1 s2.0 S0165993623001103 MainDocument12 pages1 s2.0 S0165993623001103 MainMuhammad Koksh Sdiq HussinNo ratings yet
- Ontologies in Software Testing A Systematic Literature ReviewDocument8 pagesOntologies in Software Testing A Systematic Literature ReviewafmabkbhckmajgNo ratings yet
- Molecular Docking TheoryDocument21 pagesMolecular Docking Theoryolivia6669No ratings yet
- Collision Cross Section (CCS) As A Complementary Parameter To Characterize Human and Veterinary DrugsDocument12 pagesCollision Cross Section (CCS) As A Complementary Parameter To Characterize Human and Veterinary DrugsSoraya OchsNo ratings yet
- Muegge 2010Document46 pagesMuegge 2010Santi SuronoNo ratings yet
- Dock EcrDocument10 pagesDock Ecryin chanNo ratings yet
- NOREVA - Normalization and Evaluation of MS-based Metabolomics DataDocument9 pagesNOREVA - Normalization and Evaluation of MS-based Metabolomics Dataciborg1978No ratings yet
- 2014 Biomed Res Int 348725Document13 pages2014 Biomed Res Int 348725mbrylinskiNo ratings yet
- 1 s2.0 S0010465522001175 MainDocument20 pages1 s2.0 S0010465522001175 MainI Putu Adi PratamaNo ratings yet
- DBARKHICheminf7 20Document14 pagesDBARKHICheminf7 20Risnawa Puji AstutiNo ratings yet
- A Benchmark Driven Guide To Binding SiteDocument50 pagesA Benchmark Driven Guide To Binding SiteAlexNo ratings yet
- Life Cycle Inventory Analysis: Methods and DataFrom EverandLife Cycle Inventory Analysis: Methods and DataAndreas CirothNo ratings yet
- Portfolio - Food ArticlesDocument16 pagesPortfolio - Food ArticlesDONI DERMAWANNo ratings yet
- Affinity Chromatography: Principles and Applications: Sameh Magdeldin and Annette MoserDocument26 pagesAffinity Chromatography: Principles and Applications: Sameh Magdeldin and Annette MoserDONI DERMAWANNo ratings yet
- Main-Chain Bond Lengths: ProcheckDocument1 pageMain-Chain Bond Lengths: ProcheckDONI DERMAWANNo ratings yet
- Tel: 1-631-626-9181 Fax: 1-631-614-7828Document11 pagesTel: 1-631-626-9181 Fax: 1-631-614-7828DONI DERMAWANNo ratings yet
- Main-Chain Parameters: Plot StatisticsDocument1 pageMain-Chain Parameters: Plot StatisticsDONI DERMAWANNo ratings yet
- 2019-2020 Program Guide PDFDocument142 pages2019-2020 Program Guide PDFAndhy Arya EkaputraNo ratings yet
- Biomaterials Science: Volume 2 Number 4 April 2014 Pages 429-592Document9 pagesBiomaterials Science: Volume 2 Number 4 April 2014 Pages 429-592DONI DERMAWANNo ratings yet
- System PropertyDocument3 pagesSystem Propertymoveena abdullahNo ratings yet
- Computers For DummiesDocument14 pagesComputers For Dummiesapi-256251346No ratings yet
- Janak Raikhola CVDocument3 pagesJanak Raikhola CVsaroj khanalNo ratings yet
- Course Outline PF 2021Document4 pagesCourse Outline PF 2021Kabeer UddinNo ratings yet
- Queyquep MIDTERM PRACTICESET8Document10 pagesQueyquep MIDTERM PRACTICESET8annikaNo ratings yet
- 1011202222101PM-Class 5 Revision Worksheet For First Term Examination - MathsDocument7 pages1011202222101PM-Class 5 Revision Worksheet For First Term Examination - Mathsak dNo ratings yet
- Chapter 1: Introduction (PART 1) : Computer Networking: A Top Down Approach 6th Edition Jim Kurose, Keith RossDocument15 pagesChapter 1: Introduction (PART 1) : Computer Networking: A Top Down Approach 6th Edition Jim Kurose, Keith RosssylinxNo ratings yet
- SE25 49 DAW Setup GuideDocument5 pagesSE25 49 DAW Setup GuideDiego Ignacio Garcia JuradoNo ratings yet
- Libro Criollo SaxoDocument150 pagesLibro Criollo Saxohermogenes palominoNo ratings yet
- What Is The Smart Grid? Definitions, Perspectives, and Ultimate GoalsDocument6 pagesWhat Is The Smart Grid? Definitions, Perspectives, and Ultimate GoalsAhsan TariqNo ratings yet
- Best-Network-Security ToolsDocument7 pagesBest-Network-Security Toolsesa abrenNo ratings yet
- Cyclone V Device Handbook Volume 3 Hard Processor System Technical Reference Manual PDFDocument670 pagesCyclone V Device Handbook Volume 3 Hard Processor System Technical Reference Manual PDFMaximiliano NietoNo ratings yet
- MPLS L3VPN With Sub-Interface and VRF-Lite Between PE-CE - GNS3Document11 pagesMPLS L3VPN With Sub-Interface and VRF-Lite Between PE-CE - GNS3Camps PutraNo ratings yet
- Amjad Survey PaperDocument22 pagesAmjad Survey PaperDr-Mohammed RihanNo ratings yet
- Mathmatics 1ggggggggg PDFDocument753 pagesMathmatics 1ggggggggg PDFZahid Ali Mughal100% (2)
- ReadmeDocument2 pagesReadmematheus BarbosaNo ratings yet
- ISO8583 Message Converter OnlineDocument4 pagesISO8583 Message Converter Onlinesajad salehiNo ratings yet
- Computer Shopper August 2005 PDF eBook-iNTENSiTYDocument160 pagesComputer Shopper August 2005 PDF eBook-iNTENSiTYvoodoower100% (3)
- JNTUA-B.Tech.4-1 CSE-R15-SYLLABUSDocument36 pagesJNTUA-B.Tech.4-1 CSE-R15-SYLLABUSDr William AlbertNo ratings yet
- University PUNJAB (Gujranwala Campus) : Department Information Technology Academic Transcript (Information Technology)Document2 pagesUniversity PUNJAB (Gujranwala Campus) : Department Information Technology Academic Transcript (Information Technology)HAFIZ SHAHEER UL ISLAMNo ratings yet
- Backhaul RequirementsDocument25 pagesBackhaul RequirementsChetan BhatNo ratings yet
- Lecture1113 16473 AnimationDocument11 pagesLecture1113 16473 Animationashokgehlot2100No ratings yet
- Madda Walabu University: Collage of Computing Department of Information SystemDocument22 pagesMadda Walabu University: Collage of Computing Department of Information SystemLenchoNo ratings yet
- Ethernet Cable: Color Code Standards: Basic TheoryDocument3 pagesEthernet Cable: Color Code Standards: Basic TheoryPlacid ItyNo ratings yet
- IP Com 12Document23 pagesIP Com 12Tio_louis32No ratings yet
- TEE MTCSC292 Soft Computing LabDocument2 pagesTEE MTCSC292 Soft Computing LabEKNo ratings yet
- Item ConversionDocument7 pagesItem ConversionAnu RaadhaNo ratings yet
- Dr. Öğr. Üyesi Selçuk KİZİR 1Document71 pagesDr. Öğr. Üyesi Selçuk KİZİR 1DofilomNo ratings yet