Download as pdf or txt
You might also like
- Seller Code of Conduct AppealDocument4 pagesSeller Code of Conduct AppealMAHER FAHADNo ratings yet
- Chapter - 1 Crystal Field Theory in Octahedral Complexes NotesDocument20 pagesChapter - 1 Crystal Field Theory in Octahedral Complexes NotesMohit KambojNo ratings yet
- Exciton Comsol Yao - PresentationDocument13 pagesExciton Comsol Yao - Presentationhhakim32No ratings yet
- Solar Energy Section 13 4 2Document3 pagesSolar Energy Section 13 4 2p KumarNo ratings yet
- Dielectrics 2020 FinalDocument115 pagesDielectrics 2020 FinalAninda LahiriNo ratings yet
- Spectral Switching of Type-II Quantum Dots by ChargingDocument4 pagesSpectral Switching of Type-II Quantum Dots by ChargingIndira DeviNo ratings yet
- 1 2semiconductors2Document18 pages1 2semiconductors2Muhammad HassanNo ratings yet
- Sobolev Chapter 4Document42 pagesSobolev Chapter 4UzmaNo ratings yet
- Nanostructures As Single Electron TransistorDocument16 pagesNanostructures As Single Electron TransistorM.MeenaNo ratings yet
- Electrical Engg. Dept., Sarvajanik College of Engg. and Tech., Athwalines, Surat, 390 001 Gujarat, India Elect. Engg. Dept., SVNIT, Ichchhanath, Surat, 395 007 Gujarat, IndiaDocument3 pagesElectrical Engg. Dept., Sarvajanik College of Engg. and Tech., Athwalines, Surat, 390 001 Gujarat, India Elect. Engg. Dept., SVNIT, Ichchhanath, Surat, 395 007 Gujarat, Indiacbs78No ratings yet
- (13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsDocument10 pages(13653075 - Pure and Applied Chemistry) Catalysis of Photochemical Reactions by Colloidal SemiconductorsVikash KumarNo ratings yet
- 10 1103@PhysRevB 101 081110Document5 pages10 1103@PhysRevB 101 081110Mehak MughalNo ratings yet
- Crystal Field TheoryDocument6 pagesCrystal Field TheoryRamashish ChoudharyNo ratings yet
- Song2021 Article LineDefectsInMonolayerTiSe2WitDocument6 pagesSong2021 Article LineDefectsInMonolayerTiSe2WitGonzalo FerrariNo ratings yet
- Lecture-12 CFT-1Document4 pagesLecture-12 CFT-1ABDU11AH ShafiqNo ratings yet
- Spin Flip Luminescence: Winald Robert Kitzmann Johannes Moll Katja HeinzeDocument23 pagesSpin Flip Luminescence: Winald Robert Kitzmann Johannes Moll Katja HeinzeGioele ColomboNo ratings yet
- Chapter 2-DongPanDocument28 pagesChapter 2-DongPanSri JunkNo ratings yet
- Crystal Field TheoryDocument5 pagesCrystal Field TheoryDebmalya Gharai100% (1)
- Single Electron Transistor (SET)Document8 pagesSingle Electron Transistor (SET)mokhaladNo ratings yet
- Convergence of Electronic Bands For High Performance Bulk ThermoelectricsDocument4 pagesConvergence of Electronic Bands For High Performance Bulk Thermoelectrics呂衍德No ratings yet
- Size-Dependent Energy Transfer Between Cdse/Zns Quantum Dots and Gold NanoparticlesDocument5 pagesSize-Dependent Energy Transfer Between Cdse/Zns Quantum Dots and Gold Nanoparticlessouryaya duttaNo ratings yet
- Chapter 5 (Electron Theory)Document19 pagesChapter 5 (Electron Theory)Mei Dita AsriNo ratings yet
- Rani-Tripathi2015 Article AComparativeStudyOfNanostructuDocument9 pagesRani-Tripathi2015 Article AComparativeStudyOfNanostructubismuthsunilNo ratings yet
- JP 32 91 65Document3 pagesJP 32 91 65mohamed mohamedNo ratings yet
- Struktur Pita Dan Sifat Listrik Bahan (2015.10)Document38 pagesStruktur Pita Dan Sifat Listrik Bahan (2015.10)readhybsNo ratings yet
- 49 IkeriHI 2 PDFDocument6 pages49 IkeriHI 2 PDFMohamed JaawaneNo ratings yet
- Trap Depths Analysis of Undoped KCL and Doped KCL: SRCL SystemDocument3 pagesTrap Depths Analysis of Undoped KCL and Doped KCL: SRCL SystemJournalNX - a Multidisciplinary Peer Reviewed JournalNo ratings yet
- Solid State DevicesDocument72 pagesSolid State DevicesRahul AgarwalNo ratings yet
- 2001 BESSY BorisenkoDocument2 pages2001 BESSY BorisenkoXan TolusNo ratings yet
- Single Electron Transfer Device (Setd)Document17 pagesSingle Electron Transfer Device (Setd)Krisumraj PurkaitNo ratings yet
- Densidade de Carga nbs2Document6 pagesDensidade de Carga nbs2johnfre1030No ratings yet
- Crystal Field Theory - NURDocument5 pagesCrystal Field Theory - NURNurhajrahNo ratings yet
- Dam Mak 2007Document7 pagesDam Mak 2007Daniela Araújo RodríguezNo ratings yet
- ADP013785Document12 pagesADP013785ShrabaniPaulNo ratings yet
- Solid State PhysicsDocument26 pagesSolid State PhysicskaidoqNo ratings yet
- 871 AzamSDocument13 pages871 AzamSShrabaniPaulNo ratings yet
- Temperature Effects in SemiconductorsDocument20 pagesTemperature Effects in SemiconductorsAnand ChaudharyNo ratings yet
- Solution Manual For Principles of Electronic Materials and Devices 4th Edition Kasap 0078028183 9780078028182Document7 pagesSolution Manual For Principles of Electronic Materials and Devices 4th Edition Kasap 0078028183 9780078028182anneosbornebkyomqaiz100% (30)
- Edc - Krash EceDocument75 pagesEdc - Krash EcePrabhakar DasNo ratings yet
- Magnetic Moments: Lecture 4. CHEM1902 Coordination ChemistryDocument4 pagesMagnetic Moments: Lecture 4. CHEM1902 Coordination ChemistryAminah SaqibNo ratings yet
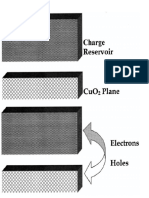
- Electronic Layers in Copper Oxide SuperconductorsDocument1 pageElectronic Layers in Copper Oxide SuperconductorsSJNo ratings yet
- Balandin NL BiTeDocument10 pagesBalandin NL BiTeShrabaniPaulNo ratings yet
- Semiconductor Lecture Notes.Document102 pagesSemiconductor Lecture Notes.bromikeseriesNo ratings yet
- Class 1-2Document40 pagesClass 1-2Aditya SukhwalNo ratings yet
- Crystal Field TheoryDocument7 pagesCrystal Field TheoryD GNo ratings yet
- Layers Crystalline Semiconductors Band Gaps HomojunctionDocument2 pagesLayers Crystalline Semiconductors Band Gaps HomojunctionLuthfiyah13No ratings yet
- 03 - Surface Tension and WettingDocument11 pages03 - Surface Tension and WettingBen PowersNo ratings yet
- Accepted Chem.201806148Document11 pagesAccepted Chem.201806148Daniela Araújo RodríguezNo ratings yet
- VBTDocument40 pagesVBTLohith LoliNo ratings yet
- Inorganic Chemistry II Second YearDocument171 pagesInorganic Chemistry II Second YearAddaa WondimeNo ratings yet
- Density of Charge Carriers in SemiconductorsDocument9 pagesDensity of Charge Carriers in SemiconductorsDasika ShishirNo ratings yet
- Observation of Dirac Charge Density Waves in Bi Te SeDocument8 pagesObservation of Dirac Charge Density Waves in Bi Te SePhysics FizicaNo ratings yet
- Molecules and Condensed Matter: Modern Physics MA in Teaching College PhysicsDocument44 pagesMolecules and Condensed Matter: Modern Physics MA in Teaching College PhysicsLogan LeeNo ratings yet
- Users Manual: Resistivity of Semiconductors by Four Probe Method at Different TemperaturesDocument23 pagesUsers Manual: Resistivity of Semiconductors by Four Probe Method at Different TemperaturesRobin ChadhaNo ratings yet
- 12 ChemDocument5 pages12 ChemBhoomi SinghNo ratings yet
- Carbon Nanotube Field-Effect Transistors and Their Possible ApplicationsDocument36 pagesCarbon Nanotube Field-Effect Transistors and Their Possible ApplicationsAkshay PatharkarNo ratings yet
- Diener 2011Document5 pagesDiener 2011johnfre1030No ratings yet
- Solution Manual For Principles of Electronic Materials and Devices 4Th Edition Kasap 0078028183 978007802818 Full Chapter PDFDocument46 pagesSolution Manual For Principles of Electronic Materials and Devices 4Th Edition Kasap 0078028183 978007802818 Full Chapter PDFkathleen.osorio990100% (18)
- Monolayer Behaviour in Bulk Res Due To Electronic and Vibrational DecouplingDocument6 pagesMonolayer Behaviour in Bulk Res Due To Electronic and Vibrational DecouplingMario Flores SalazarNo ratings yet
- Coatings 13 00208Document12 pagesCoatings 13 00208Mira SucheaNo ratings yet
- Copper Zinc Tin Sulfide-Based Thin-Film Solar CellsFrom EverandCopper Zinc Tin Sulfide-Based Thin-Film Solar CellsKentaro ItoNo ratings yet
- Technical Data Sheet: Tire Lube and SealDocument1 pageTechnical Data Sheet: Tire Lube and SealDon HowardNo ratings yet
- Maintenance Manual 1T-4T (LPG) - EN 英语Document128 pagesMaintenance Manual 1T-4T (LPG) - EN 英语Jose Luis LavinNo ratings yet
- Phy Investigatory Project Tangent GalvanDocument11 pagesPhy Investigatory Project Tangent Galvankomal mahelaNo ratings yet
- Comprehensive Report Card: About The Problems of Sub-Health TrendsDocument3 pagesComprehensive Report Card: About The Problems of Sub-Health TrendsFitriWidiastutiNo ratings yet
- IS0125 F Fuel-ManagerDocument12 pagesIS0125 F Fuel-ManagerBilly12369No ratings yet
- Programme Title:: (Dd/mm/yyyy) (XXX)Document4 pagesProgramme Title:: (Dd/mm/yyyy) (XXX)ghanNo ratings yet
- Requiem Bloodlines & PowersDocument16 pagesRequiem Bloodlines & PowersLuiz Henrique Matias MarcondesNo ratings yet
- Rock Mechanics Fundamentals: Foot) - What Is Its Specific Gravity?Document18 pagesRock Mechanics Fundamentals: Foot) - What Is Its Specific Gravity?Ghulam Mohyuddin SohailNo ratings yet
- AC Joint Reconstruction ProtocolDocument20 pagesAC Joint Reconstruction ProtocolAnomalie12345No ratings yet
- The Basics of Arc WeldingDocument35 pagesThe Basics of Arc WeldingDesmond ChangNo ratings yet
- Effects of Experimental Parameters On NF3 Decomposition Fraction in An Oxygen-Based 2004Document7 pagesEffects of Experimental Parameters On NF3 Decomposition Fraction in An Oxygen-Based 2004Регина ШаяхметоваNo ratings yet
- BrainDocument41 pagesBrainNishanth Siva100% (1)
- TR1 G-Build Technical Data SheetDocument2 pagesTR1 G-Build Technical Data SheetHernan B.No ratings yet
- Code On Wages 2019 - NotesDocument3 pagesCode On Wages 2019 - NotesAnand ReddyNo ratings yet
- 12 Production and Purification of Recombinant Glargine Insulin From Escherichia Coli BL-21 StrainDocument12 pages12 Production and Purification of Recombinant Glargine Insulin From Escherichia Coli BL-21 StrainAnand KumarNo ratings yet
- Pressure Conversion ChartDocument1 pagePressure Conversion Chartamarkore3486No ratings yet
- Ebook Encyclopedia of Cardiovascular Research and Medicine PDF Full Chapter PDFDocument47 pagesEbook Encyclopedia of Cardiovascular Research and Medicine PDF Full Chapter PDFkimberly.dixon591100% (30)
- Case Ih Tractor 1070 Operators Manual 9 2822Document22 pagesCase Ih Tractor 1070 Operators Manual 9 2822zacharygonzalez130703jse100% (140)
- Condenser Sizing CalculationDocument21 pagesCondenser Sizing CalculationShruti Sharma100% (4)
- Attachment 15 The Silent Genocide of The Boer Nation in South Africa IndexDocument3 pagesAttachment 15 The Silent Genocide of The Boer Nation in South Africa Indexapi-232649836No ratings yet
- Nutrient Ciclagem de NutrientesDocument352 pagesNutrient Ciclagem de NutrientesFabio SouzaNo ratings yet
- Crankcase Blowby, Measure PDF FORMATDocument51 pagesCrankcase Blowby, Measure PDF FORMATDARISON VINCENT100% (2)
- Drexel SL 30-40-50 AC MM F-626-0419Document140 pagesDrexel SL 30-40-50 AC MM F-626-0419Abel GonzalezNo ratings yet
- Call For Applications For PHD in Midwifery September 2023 Intake 6.6.23Document3 pagesCall For Applications For PHD in Midwifery September 2023 Intake 6.6.23EvansNo ratings yet
- Lecture 4Document26 pagesLecture 4shahad mNo ratings yet
- Huizhe Wu, MD Mingyan Liu, MD Shuang Wang, MD Wanyu Feng, MD, PHD Weifan Yao, Bs Haishan Zhao, Bs and Minjie Wei, MD, PHDDocument10 pagesHuizhe Wu, MD Mingyan Liu, MD Shuang Wang, MD Wanyu Feng, MD, PHD Weifan Yao, Bs Haishan Zhao, Bs and Minjie Wei, MD, PHDDyva VanillaNo ratings yet
- Module 2Document40 pagesModule 2Romel CuregNo ratings yet
- BacktoBasics-Fundamental Resolution Equation V2Document7 pagesBacktoBasics-Fundamental Resolution Equation V2Yeoh XWNo ratings yet