
1982-Joc-3397, 2-Iodooctane
1982-Joc-3397, 2-Iodooctane
You might also like
- Cheat SheetDocument5 pagesCheat Sheetkittenface92% (13)
- 19 TVR6G ID+OD 50Hz Product CatalgoueDocument98 pages19 TVR6G ID+OD 50Hz Product CatalgoueKareem Abo Seif100% (1)
- Formal Report On Enzymes: Effect of PH and Temperature On Invertase ActivityDocument4 pagesFormal Report On Enzymes: Effect of PH and Temperature On Invertase ActivityYoreeNo ratings yet
- Electrochemical Oxidation of Benzoic Acid at Boron-Doped Diamond ElectrodesDocument5 pagesElectrochemical Oxidation of Benzoic Acid at Boron-Doped Diamond ElectrodesSebas ValenciaNo ratings yet
- C 3 RedoxDocument28 pagesC 3 RedoxRina P ShresthaNo ratings yet
- Roto 2002Document8 pagesRoto 2002k.suganeswaranNo ratings yet
- Mathematical Modelling and Simulation AnDocument7 pagesMathematical Modelling and Simulation AnHusamZarourNo ratings yet
- Study of The Underlying Electrochemistry of Polycrystalline GoldDocument13 pagesStudy of The Underlying Electrochemistry of Polycrystalline GoldAzucena osornio villaNo ratings yet
- Mete Alp Yıldırım EXP 10 ReportDocument7 pagesMete Alp Yıldırım EXP 10 ReportAlp YıldırımNo ratings yet
- Electrolyzer Modules A) Unipolar and B) Bipolar Cell ConfigurationsDocument8 pagesElectrolyzer Modules A) Unipolar and B) Bipolar Cell ConfigurationsAnuj ShahiNo ratings yet
- A Review On Water ElectrolysisDocument18 pagesA Review On Water ElectrolysisSilvester KolicNo ratings yet
- Electrochemical Oxidation / Reduction: Physicochemical Processes ClassDocument48 pagesElectrochemical Oxidation / Reduction: Physicochemical Processes ClassErnest NsabimanaNo ratings yet
- Electrolysis LessonDocument25 pagesElectrolysis LessonAbigail MedinaNo ratings yet
- PH.D - Synopsis - M. Praveen KumarDocument12 pagesPH.D - Synopsis - M. Praveen KumaralexabcdxyzNo ratings yet
- Study of The Electrocoagulation of Electroplating Industry Wastewaters Charged by Nickel (II) and Chromium (VI)Document10 pagesStudy of The Electrocoagulation of Electroplating Industry Wastewaters Charged by Nickel (II) and Chromium (VI)Martin FernandezNo ratings yet
- Chemistry and Electricity:: ElectrochemistryDocument5 pagesChemistry and Electricity:: ElectrochemistrySuleman TariqNo ratings yet
- Electrochemical Degradation of Bromopyrogallol Red in Presence of Cobalt IonsDocument6 pagesElectrochemical Degradation of Bromopyrogallol Red in Presence of Cobalt Ionsapi-3828788No ratings yet
- Electrocatalysis in Wastewater Treatment Recent MeDocument9 pagesElectrocatalysis in Wastewater Treatment Recent MeSebastian FNo ratings yet
- Art Sal TantalioDocument6 pagesArt Sal TantalioJavier Mauricio Neira CastrillonNo ratings yet
- Lecture Notes - Engineering ChemistryDocument56 pagesLecture Notes - Engineering ChemistryadamjosephNo ratings yet
- Electrolytic ConductionDocument38 pagesElectrolytic ConductionVishwanath ReddyNo ratings yet
- Module 1 - Electrochemistry (Part 1)Document11 pagesModule 1 - Electrochemistry (Part 1)Steven Lee100% (2)
- Carbon Film Electrodes As Support of Metallic Particles: Int. J. Electrochem. Sci., 7 (2012) 150 - 166Document17 pagesCarbon Film Electrodes As Support of Metallic Particles: Int. J. Electrochem. Sci., 7 (2012) 150 - 166Felipe Scholz RamosNo ratings yet
- Module 1 - ElectrochemistryDocument31 pagesModule 1 - ElectrochemistryjeniferNo ratings yet
- 12 Chemistry Impq CH03 Electro Chemistry 02 PDFDocument9 pages12 Chemistry Impq CH03 Electro Chemistry 02 PDFamanNo ratings yet
- 12 Chemistry Impq CH03 Electro Chemistry 02 PDFDocument9 pages12 Chemistry Impq CH03 Electro Chemistry 02 PDFamanNo ratings yet
- Corrosion: Chap. 2Document53 pagesCorrosion: Chap. 2Daniel RomeroNo ratings yet
- Anodic Reactions: 08Y506 - Corrosion EngineeringDocument5 pagesAnodic Reactions: 08Y506 - Corrosion Engineeringesakkibabu1987No ratings yet
- The Direct Electrochemical Synthesis of ( (C6H5) 3Ph) 2 (CoCl4)Document2 pagesThe Direct Electrochemical Synthesis of ( (C6H5) 3Ph) 2 (CoCl4)Pavle RadojkovićNo ratings yet
- 1 s2.0 S0360319901001598 Main PDFDocument6 pages1 s2.0 S0360319901001598 Main PDFDr ChNo ratings yet
- Electrochemistry: (Tuesday, 8 May 2017)Document18 pagesElectrochemistry: (Tuesday, 8 May 2017)mipa amarNo ratings yet
- Water ElectrolysisDocument4 pagesWater ElectrolysisScribdTranslationsNo ratings yet
- Unit 12 ElectrochemistryDocument22 pagesUnit 12 Electrochemistrycream oNo ratings yet
- CMT555-1-Electrochemical Cells & Thermodynamics-Stdnt NotesDocument72 pagesCMT555-1-Electrochemical Cells & Thermodynamics-Stdnt NotesjuaxxoNo ratings yet
- Electrolysis: Experiment 5Document9 pagesElectrolysis: Experiment 5ari wiliamNo ratings yet
- 1158 1 OnlineDocument10 pages1158 1 OnlineSebastián Alberto Campos MillaNo ratings yet
- Electrolysis of Water - WikipediaDocument20 pagesElectrolysis of Water - Wikipediapowew28978No ratings yet
- Relationship Between The Morphology For The Photo-Electrode of Copper Bismuth Oxide and The Photo-Electrochemical Activity Related To Water ReductionDocument10 pagesRelationship Between The Morphology For The Photo-Electrode of Copper Bismuth Oxide and The Photo-Electrochemical Activity Related To Water ReductionmuhammadNo ratings yet
- This Article Is About The Chemical Process. For The Cosmetic Hair Removal Procedure, SeeDocument4 pagesThis Article Is About The Chemical Process. For The Cosmetic Hair Removal Procedure, Seewaseem555No ratings yet
- IB HL Chemistry Assessment Statements Topics 9 and 19Document4 pagesIB HL Chemistry Assessment Statements Topics 9 and 19AndrewNo ratings yet
- CH 3 14Document135 pagesCH 3 14active learning educationNo ratings yet
- Redox Reaction: Oxidation and Reduction in Terms of Oxygen TransferDocument28 pagesRedox Reaction: Oxidation and Reduction in Terms of Oxygen TransferfaridNo ratings yet
- Water: Water Radiolysis: Influence of Oxide Surfaces On H Production Under Ionizing RadiationDocument19 pagesWater: Water Radiolysis: Influence of Oxide Surfaces On H Production Under Ionizing RadiationSeptiana SariNo ratings yet
- Catalysts 09 00243Document14 pagesCatalysts 09 00243Ghirlie Eunice LopezNo ratings yet
- Unit 2 Electrochemistry and Energy Storage SystemsDocument24 pagesUnit 2 Electrochemistry and Energy Storage Systemsudhayakumar7848No ratings yet
- Oxidación Bioelectroquímica Del AguaDocument4 pagesOxidación Bioelectroquímica Del AguaGabriela CervantesNo ratings yet
- Chem 17Document9 pagesChem 17Adi SoNo ratings yet
- Plasmonic Photo Catalyst For H2 Evolution in Photo Catalytic Water SplittingDocument7 pagesPlasmonic Photo Catalyst For H2 Evolution in Photo Catalytic Water Splittingbsnyder3No ratings yet
- QR 9672100079Document30 pagesQR 9672100079MycoLogist4LifeNo ratings yet
- Chem 2Document15 pagesChem 2CR7STUDIO 70% (1)
- Lesson 1 FunctionsDocument7 pagesLesson 1 FunctionsWaien G. WatamamaNo ratings yet
- MODULE 2 ElectrochemistryDocument31 pagesMODULE 2 ElectrochemistryChristian Mark De JesusNo ratings yet
- ElectrochemistryDocument26 pagesElectrochemistryJasbir SinghNo ratings yet
- General Chemistry 2 Quarter 4: Week 7 - Module 7 Standard Cell Potential, Electrochemical Cells and BatteriesDocument21 pagesGeneral Chemistry 2 Quarter 4: Week 7 - Module 7 Standard Cell Potential, Electrochemical Cells and BatteriesCamille Joves EncarnacionNo ratings yet
- Laboratory Study of Electro-Coagulation - Otation For Water TreatmentDocument15 pagesLaboratory Study of Electro-Coagulation - Otation For Water TreatmentazerfazNo ratings yet
- Molecule-Independent Electrical Switching in PT Organic Monolayer Ti DevicesDocument4 pagesMolecule-Independent Electrical Switching in PT Organic Monolayer Ti Devicesraymond wellNo ratings yet
- Basic Corrosion Theory: 2.1 ThermodynamicsDocument2 pagesBasic Corrosion Theory: 2.1 Thermodynamicssri ramadhaniNo ratings yet
- Concept of ElectrochemitryDocument15 pagesConcept of ElectrochemitryKritika SainiNo ratings yet
- Sustainable and Green Electrochemical Science and TechnologyFrom EverandSustainable and Green Electrochemical Science and TechnologyNo ratings yet
- Inorganic Reactions and Methods, Electron-Transfer and Electrochemical Reactions; Photochemical and Other Energized ReactionsFrom EverandInorganic Reactions and Methods, Electron-Transfer and Electrochemical Reactions; Photochemical and Other Energized ReactionsNo ratings yet
- J Jelechem 2017 12 013Document5 pagesJ Jelechem 2017 12 013محمد مصطفىNo ratings yet
- SFP PDFDocument12 pagesSFP PDFمحمد مصطفى100% (1)
- Su Slick 1990Document8 pagesSu Slick 1990محمد مصطفىNo ratings yet
- Advanced Organic Chemistry/ Organic Synthesis - CH 621 Ultrasound Assisted Organic Synthesis (Sonochemistry)Document36 pagesAdvanced Organic Chemistry/ Organic Synthesis - CH 621 Ultrasound Assisted Organic Synthesis (Sonochemistry)محمد مصطفىNo ratings yet
- And Finally: Jeffrey I. SeemanDocument16 pagesAnd Finally: Jeffrey I. Seemanمحمد مصطفىNo ratings yet
- Diazo Synthesis ImportantDocument17 pagesDiazo Synthesis Importantمحمد مصطفىNo ratings yet
- Double GroupDocument11 pagesDouble Groupمحمد مصطفىNo ratings yet
- Synthesis of Tropinone & 2-CMT, Hive Methods DiscourseDocument10 pagesSynthesis of Tropinone & 2-CMT, Hive Methods Discourseمحمد مصطفىNo ratings yet
- Synthetic Organic Chemistry 932 Jalal Zahra Homework 1Document1 pageSynthetic Organic Chemistry 932 Jalal Zahra Homework 1محمد مصطفىNo ratings yet
- SS 5100aDocument2 pagesSS 5100aKannan Krishnamurthy100% (1)
- Mineral Processing A DasDocument36 pagesMineral Processing A DasYallarling NagureNo ratings yet
- EAT203 - Lab ReportDocument24 pagesEAT203 - Lab ReportRavi VarmanNo ratings yet
- Structure of Flexible PackagingDocument179 pagesStructure of Flexible Packagingkam ka weiNo ratings yet
- Product - Info High Energy Ball Mill Emax ENDocument6 pagesProduct - Info High Energy Ball Mill Emax ENaditya triatmajaNo ratings yet
- 004 Calculating ImpedanceDocument5 pages004 Calculating ImpedancedaddadNo ratings yet
- S2 Covid-19 Holiday Work Week Two PDFDocument3 pagesS2 Covid-19 Holiday Work Week Two PDFLutwama GideonNo ratings yet
- Assignment - 1 (Structure of Atom)Document2 pagesAssignment - 1 (Structure of Atom)gobinda prasad barmanNo ratings yet
- Augmented Pool Boiling Heat Transfer of PF-5060 Using Femtosecond Laser Surface Processed Aluminum Effect of Laser FluenceDocument9 pagesAugmented Pool Boiling Heat Transfer of PF-5060 Using Femtosecond Laser Surface Processed Aluminum Effect of Laser Fluencenitish kumarNo ratings yet
- E-Program Files-AN-ConnectManager-SSIS-MSDS-PDF-HFA062 - NZ - EN - 20160512 - 1Document10 pagesE-Program Files-AN-ConnectManager-SSIS-MSDS-PDF-HFA062 - NZ - EN - 20160512 - 1Alex MaduNo ratings yet
- DLL September 4 7Document3 pagesDLL September 4 7DENVER MOSCOSONo ratings yet
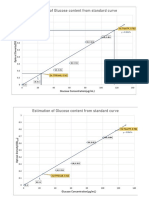
- Estimation of Glucose Content From Standard Curve: Glucose Concentration ( G/ML)Document1 pageEstimation of Glucose Content From Standard Curve: Glucose Concentration ( G/ML)Hasan AnandaNo ratings yet
- Design and Construction of A Water Scrubber For The Upgrading of BiogasDocument6 pagesDesign and Construction of A Water Scrubber For The Upgrading of BiogasPreetham BharadwajNo ratings yet
- Ssags PSV SizingDocument5 pagesSsags PSV SizingEkundayo JohnNo ratings yet
- TT M.Sc. I SEM (C.B.C.S.) 21012020Document2 pagesTT M.Sc. I SEM (C.B.C.S.) 21012020Akash RautNo ratings yet
- Practice 7Document3 pagesPractice 7Neha SharmaNo ratings yet
- GenbioDocument5 pagesGenbioYkhay ElfanteNo ratings yet
- Assignment 02 - IPE2230 3Document4 pagesAssignment 02 - IPE2230 3MD Al-AminNo ratings yet
- Parsol 1789Document20 pagesParsol 1789Dr.Vinod SrivastavaNo ratings yet
- Formulation and Evaluation of Floating and Mucoadhesive Tablets Containing GlipizideDocument8 pagesFormulation and Evaluation of Floating and Mucoadhesive Tablets Containing GlipizideSiva PrasadNo ratings yet
- Formula Sheet: MFM 2P: W L P W L P LW ADocument2 pagesFormula Sheet: MFM 2P: W L P W L P LW AKIMIA PARNIANINo ratings yet
- Frames of Reference: Ion PropulsionDocument3 pagesFrames of Reference: Ion PropulsionLakshyaNo ratings yet
- Static Compaction Test and Determination of Equivalent Static PressureDocument4 pagesStatic Compaction Test and Determination of Equivalent Static PressureRkNo ratings yet
- Guide For DLS Sample Preparation: Eric Farrell & Jean-Luc Brousseau PH.DDocument3 pagesGuide For DLS Sample Preparation: Eric Farrell & Jean-Luc Brousseau PH.DDeidre CadeNo ratings yet
- IOM - Gas Detector - AE - AIYI - EN Manual-AG310 Series Fixed Gas DetectorDocument28 pagesIOM - Gas Detector - AE - AIYI - EN Manual-AG310 Series Fixed Gas DetectoramielNo ratings yet
- Power CEPDocument5 pagesPower CEPhamzadaud032No ratings yet
- Beta and Gamma RaysDocument10 pagesBeta and Gamma RaysEnoch SpalbarNo ratings yet
- Technical Data Sheet: Survey of GradesDocument12 pagesTechnical Data Sheet: Survey of GradesOmkarNo ratings yet


































































































Download as pdf or txt
You might also like
- Cheat SheetDocument5 pagesCheat Sheetkittenface92% (13)
- 19 TVR6G ID+OD 50Hz Product CatalgoueDocument98 pages19 TVR6G ID+OD 50Hz Product CatalgoueKareem Abo Seif100% (1)
- Formal Report On Enzymes: Effect of PH and Temperature On Invertase ActivityDocument4 pagesFormal Report On Enzymes: Effect of PH and Temperature On Invertase ActivityYoreeNo ratings yet
- Electrochemical Oxidation of Benzoic Acid at Boron-Doped Diamond ElectrodesDocument5 pagesElectrochemical Oxidation of Benzoic Acid at Boron-Doped Diamond ElectrodesSebas ValenciaNo ratings yet
- C 3 RedoxDocument28 pagesC 3 RedoxRina P ShresthaNo ratings yet
- Roto 2002Document8 pagesRoto 2002k.suganeswaranNo ratings yet
- Mathematical Modelling and Simulation AnDocument7 pagesMathematical Modelling and Simulation AnHusamZarourNo ratings yet
- Study of The Underlying Electrochemistry of Polycrystalline GoldDocument13 pagesStudy of The Underlying Electrochemistry of Polycrystalline GoldAzucena osornio villaNo ratings yet
- Mete Alp Yıldırım EXP 10 ReportDocument7 pagesMete Alp Yıldırım EXP 10 ReportAlp YıldırımNo ratings yet
- Electrolyzer Modules A) Unipolar and B) Bipolar Cell ConfigurationsDocument8 pagesElectrolyzer Modules A) Unipolar and B) Bipolar Cell ConfigurationsAnuj ShahiNo ratings yet
- A Review On Water ElectrolysisDocument18 pagesA Review On Water ElectrolysisSilvester KolicNo ratings yet
- Electrochemical Oxidation / Reduction: Physicochemical Processes ClassDocument48 pagesElectrochemical Oxidation / Reduction: Physicochemical Processes ClassErnest NsabimanaNo ratings yet
- Electrolysis LessonDocument25 pagesElectrolysis LessonAbigail MedinaNo ratings yet
- PH.D - Synopsis - M. Praveen KumarDocument12 pagesPH.D - Synopsis - M. Praveen KumaralexabcdxyzNo ratings yet
- Study of The Electrocoagulation of Electroplating Industry Wastewaters Charged by Nickel (II) and Chromium (VI)Document10 pagesStudy of The Electrocoagulation of Electroplating Industry Wastewaters Charged by Nickel (II) and Chromium (VI)Martin FernandezNo ratings yet
- Chemistry and Electricity:: ElectrochemistryDocument5 pagesChemistry and Electricity:: ElectrochemistrySuleman TariqNo ratings yet
- Electrochemical Degradation of Bromopyrogallol Red in Presence of Cobalt IonsDocument6 pagesElectrochemical Degradation of Bromopyrogallol Red in Presence of Cobalt Ionsapi-3828788No ratings yet
- Electrocatalysis in Wastewater Treatment Recent MeDocument9 pagesElectrocatalysis in Wastewater Treatment Recent MeSebastian FNo ratings yet
- Art Sal TantalioDocument6 pagesArt Sal TantalioJavier Mauricio Neira CastrillonNo ratings yet
- Lecture Notes - Engineering ChemistryDocument56 pagesLecture Notes - Engineering ChemistryadamjosephNo ratings yet
- Electrolytic ConductionDocument38 pagesElectrolytic ConductionVishwanath ReddyNo ratings yet
- Module 1 - Electrochemistry (Part 1)Document11 pagesModule 1 - Electrochemistry (Part 1)Steven Lee100% (2)
- Carbon Film Electrodes As Support of Metallic Particles: Int. J. Electrochem. Sci., 7 (2012) 150 - 166Document17 pagesCarbon Film Electrodes As Support of Metallic Particles: Int. J. Electrochem. Sci., 7 (2012) 150 - 166Felipe Scholz RamosNo ratings yet
- Module 1 - ElectrochemistryDocument31 pagesModule 1 - ElectrochemistryjeniferNo ratings yet
- 12 Chemistry Impq CH03 Electro Chemistry 02 PDFDocument9 pages12 Chemistry Impq CH03 Electro Chemistry 02 PDFamanNo ratings yet
- 12 Chemistry Impq CH03 Electro Chemistry 02 PDFDocument9 pages12 Chemistry Impq CH03 Electro Chemistry 02 PDFamanNo ratings yet
- Corrosion: Chap. 2Document53 pagesCorrosion: Chap. 2Daniel RomeroNo ratings yet
- Anodic Reactions: 08Y506 - Corrosion EngineeringDocument5 pagesAnodic Reactions: 08Y506 - Corrosion Engineeringesakkibabu1987No ratings yet
- The Direct Electrochemical Synthesis of ( (C6H5) 3Ph) 2 (CoCl4)Document2 pagesThe Direct Electrochemical Synthesis of ( (C6H5) 3Ph) 2 (CoCl4)Pavle RadojkovićNo ratings yet
- 1 s2.0 S0360319901001598 Main PDFDocument6 pages1 s2.0 S0360319901001598 Main PDFDr ChNo ratings yet
- Electrochemistry: (Tuesday, 8 May 2017)Document18 pagesElectrochemistry: (Tuesday, 8 May 2017)mipa amarNo ratings yet
- Water ElectrolysisDocument4 pagesWater ElectrolysisScribdTranslationsNo ratings yet
- Unit 12 ElectrochemistryDocument22 pagesUnit 12 Electrochemistrycream oNo ratings yet
- CMT555-1-Electrochemical Cells & Thermodynamics-Stdnt NotesDocument72 pagesCMT555-1-Electrochemical Cells & Thermodynamics-Stdnt NotesjuaxxoNo ratings yet
- Electrolysis: Experiment 5Document9 pagesElectrolysis: Experiment 5ari wiliamNo ratings yet
- 1158 1 OnlineDocument10 pages1158 1 OnlineSebastián Alberto Campos MillaNo ratings yet
- Electrolysis of Water - WikipediaDocument20 pagesElectrolysis of Water - Wikipediapowew28978No ratings yet
- Relationship Between The Morphology For The Photo-Electrode of Copper Bismuth Oxide and The Photo-Electrochemical Activity Related To Water ReductionDocument10 pagesRelationship Between The Morphology For The Photo-Electrode of Copper Bismuth Oxide and The Photo-Electrochemical Activity Related To Water ReductionmuhammadNo ratings yet
- This Article Is About The Chemical Process. For The Cosmetic Hair Removal Procedure, SeeDocument4 pagesThis Article Is About The Chemical Process. For The Cosmetic Hair Removal Procedure, Seewaseem555No ratings yet
- IB HL Chemistry Assessment Statements Topics 9 and 19Document4 pagesIB HL Chemistry Assessment Statements Topics 9 and 19AndrewNo ratings yet
- CH 3 14Document135 pagesCH 3 14active learning educationNo ratings yet
- Redox Reaction: Oxidation and Reduction in Terms of Oxygen TransferDocument28 pagesRedox Reaction: Oxidation and Reduction in Terms of Oxygen TransferfaridNo ratings yet
- Water: Water Radiolysis: Influence of Oxide Surfaces On H Production Under Ionizing RadiationDocument19 pagesWater: Water Radiolysis: Influence of Oxide Surfaces On H Production Under Ionizing RadiationSeptiana SariNo ratings yet
- Catalysts 09 00243Document14 pagesCatalysts 09 00243Ghirlie Eunice LopezNo ratings yet
- Unit 2 Electrochemistry and Energy Storage SystemsDocument24 pagesUnit 2 Electrochemistry and Energy Storage Systemsudhayakumar7848No ratings yet
- Oxidación Bioelectroquímica Del AguaDocument4 pagesOxidación Bioelectroquímica Del AguaGabriela CervantesNo ratings yet
- Chem 17Document9 pagesChem 17Adi SoNo ratings yet
- Plasmonic Photo Catalyst For H2 Evolution in Photo Catalytic Water SplittingDocument7 pagesPlasmonic Photo Catalyst For H2 Evolution in Photo Catalytic Water Splittingbsnyder3No ratings yet
- QR 9672100079Document30 pagesQR 9672100079MycoLogist4LifeNo ratings yet
- Chem 2Document15 pagesChem 2CR7STUDIO 70% (1)
- Lesson 1 FunctionsDocument7 pagesLesson 1 FunctionsWaien G. WatamamaNo ratings yet
- MODULE 2 ElectrochemistryDocument31 pagesMODULE 2 ElectrochemistryChristian Mark De JesusNo ratings yet
- ElectrochemistryDocument26 pagesElectrochemistryJasbir SinghNo ratings yet
- General Chemistry 2 Quarter 4: Week 7 - Module 7 Standard Cell Potential, Electrochemical Cells and BatteriesDocument21 pagesGeneral Chemistry 2 Quarter 4: Week 7 - Module 7 Standard Cell Potential, Electrochemical Cells and BatteriesCamille Joves EncarnacionNo ratings yet
- Laboratory Study of Electro-Coagulation - Otation For Water TreatmentDocument15 pagesLaboratory Study of Electro-Coagulation - Otation For Water TreatmentazerfazNo ratings yet
- Molecule-Independent Electrical Switching in PT Organic Monolayer Ti DevicesDocument4 pagesMolecule-Independent Electrical Switching in PT Organic Monolayer Ti Devicesraymond wellNo ratings yet
- Basic Corrosion Theory: 2.1 ThermodynamicsDocument2 pagesBasic Corrosion Theory: 2.1 Thermodynamicssri ramadhaniNo ratings yet
- Concept of ElectrochemitryDocument15 pagesConcept of ElectrochemitryKritika SainiNo ratings yet
- Sustainable and Green Electrochemical Science and TechnologyFrom EverandSustainable and Green Electrochemical Science and TechnologyNo ratings yet
- Inorganic Reactions and Methods, Electron-Transfer and Electrochemical Reactions; Photochemical and Other Energized ReactionsFrom EverandInorganic Reactions and Methods, Electron-Transfer and Electrochemical Reactions; Photochemical and Other Energized ReactionsNo ratings yet
- J Jelechem 2017 12 013Document5 pagesJ Jelechem 2017 12 013محمد مصطفىNo ratings yet
- SFP PDFDocument12 pagesSFP PDFمحمد مصطفى100% (1)
- Su Slick 1990Document8 pagesSu Slick 1990محمد مصطفىNo ratings yet
- Advanced Organic Chemistry/ Organic Synthesis - CH 621 Ultrasound Assisted Organic Synthesis (Sonochemistry)Document36 pagesAdvanced Organic Chemistry/ Organic Synthesis - CH 621 Ultrasound Assisted Organic Synthesis (Sonochemistry)محمد مصطفىNo ratings yet
- And Finally: Jeffrey I. SeemanDocument16 pagesAnd Finally: Jeffrey I. Seemanمحمد مصطفىNo ratings yet
- Diazo Synthesis ImportantDocument17 pagesDiazo Synthesis Importantمحمد مصطفىNo ratings yet
- Double GroupDocument11 pagesDouble Groupمحمد مصطفىNo ratings yet
- Synthesis of Tropinone & 2-CMT, Hive Methods DiscourseDocument10 pagesSynthesis of Tropinone & 2-CMT, Hive Methods Discourseمحمد مصطفىNo ratings yet
- Synthetic Organic Chemistry 932 Jalal Zahra Homework 1Document1 pageSynthetic Organic Chemistry 932 Jalal Zahra Homework 1محمد مصطفىNo ratings yet
- SS 5100aDocument2 pagesSS 5100aKannan Krishnamurthy100% (1)
- Mineral Processing A DasDocument36 pagesMineral Processing A DasYallarling NagureNo ratings yet
- EAT203 - Lab ReportDocument24 pagesEAT203 - Lab ReportRavi VarmanNo ratings yet
- Structure of Flexible PackagingDocument179 pagesStructure of Flexible Packagingkam ka weiNo ratings yet
- Product - Info High Energy Ball Mill Emax ENDocument6 pagesProduct - Info High Energy Ball Mill Emax ENaditya triatmajaNo ratings yet
- 004 Calculating ImpedanceDocument5 pages004 Calculating ImpedancedaddadNo ratings yet
- S2 Covid-19 Holiday Work Week Two PDFDocument3 pagesS2 Covid-19 Holiday Work Week Two PDFLutwama GideonNo ratings yet
- Assignment - 1 (Structure of Atom)Document2 pagesAssignment - 1 (Structure of Atom)gobinda prasad barmanNo ratings yet
- Augmented Pool Boiling Heat Transfer of PF-5060 Using Femtosecond Laser Surface Processed Aluminum Effect of Laser FluenceDocument9 pagesAugmented Pool Boiling Heat Transfer of PF-5060 Using Femtosecond Laser Surface Processed Aluminum Effect of Laser Fluencenitish kumarNo ratings yet
- E-Program Files-AN-ConnectManager-SSIS-MSDS-PDF-HFA062 - NZ - EN - 20160512 - 1Document10 pagesE-Program Files-AN-ConnectManager-SSIS-MSDS-PDF-HFA062 - NZ - EN - 20160512 - 1Alex MaduNo ratings yet
- DLL September 4 7Document3 pagesDLL September 4 7DENVER MOSCOSONo ratings yet
- Estimation of Glucose Content From Standard Curve: Glucose Concentration ( G/ML)Document1 pageEstimation of Glucose Content From Standard Curve: Glucose Concentration ( G/ML)Hasan AnandaNo ratings yet
- Design and Construction of A Water Scrubber For The Upgrading of BiogasDocument6 pagesDesign and Construction of A Water Scrubber For The Upgrading of BiogasPreetham BharadwajNo ratings yet
- Ssags PSV SizingDocument5 pagesSsags PSV SizingEkundayo JohnNo ratings yet
- TT M.Sc. I SEM (C.B.C.S.) 21012020Document2 pagesTT M.Sc. I SEM (C.B.C.S.) 21012020Akash RautNo ratings yet
- Practice 7Document3 pagesPractice 7Neha SharmaNo ratings yet
- GenbioDocument5 pagesGenbioYkhay ElfanteNo ratings yet
- Assignment 02 - IPE2230 3Document4 pagesAssignment 02 - IPE2230 3MD Al-AminNo ratings yet
- Parsol 1789Document20 pagesParsol 1789Dr.Vinod SrivastavaNo ratings yet
- Formulation and Evaluation of Floating and Mucoadhesive Tablets Containing GlipizideDocument8 pagesFormulation and Evaluation of Floating and Mucoadhesive Tablets Containing GlipizideSiva PrasadNo ratings yet
- Formula Sheet: MFM 2P: W L P W L P LW ADocument2 pagesFormula Sheet: MFM 2P: W L P W L P LW AKIMIA PARNIANINo ratings yet
- Frames of Reference: Ion PropulsionDocument3 pagesFrames of Reference: Ion PropulsionLakshyaNo ratings yet
- Static Compaction Test and Determination of Equivalent Static PressureDocument4 pagesStatic Compaction Test and Determination of Equivalent Static PressureRkNo ratings yet
- Guide For DLS Sample Preparation: Eric Farrell & Jean-Luc Brousseau PH.DDocument3 pagesGuide For DLS Sample Preparation: Eric Farrell & Jean-Luc Brousseau PH.DDeidre CadeNo ratings yet
- IOM - Gas Detector - AE - AIYI - EN Manual-AG310 Series Fixed Gas DetectorDocument28 pagesIOM - Gas Detector - AE - AIYI - EN Manual-AG310 Series Fixed Gas DetectoramielNo ratings yet
- Power CEPDocument5 pagesPower CEPhamzadaud032No ratings yet
- Beta and Gamma RaysDocument10 pagesBeta and Gamma RaysEnoch SpalbarNo ratings yet
- Technical Data Sheet: Survey of GradesDocument12 pagesTechnical Data Sheet: Survey of GradesOmkarNo ratings yet