Download as docx, pdf, or txt
You might also like
- REG.8.2-01-03 Post-Market - Surveillance - ReportDocument4 pagesREG.8.2-01-03 Post-Market - Surveillance - ReportMarcBenetPozo100% (10)
- ISO TR 20416-2020 Medical Devices-Post-Market Surveillance For ManufacturersDocument50 pagesISO TR 20416-2020 Medical Devices-Post-Market Surveillance For ManufacturersQas Wei100% (4)
- REG.8.2-01-00 Post-Market - Surveillance - Plan - # - 2019 - 02 - 04 - # - V2Document4 pagesREG.8.2-01-00 Post-Market - Surveillance - Plan - # - 2019 - 02 - 04 - # - V2MarcBenetPozo100% (1)
- FDA Medical Device Complaint FormDocument2 pagesFDA Medical Device Complaint Formclayton51175% (4)
- Standard Operating Procedure For Key Peformance IndicatorsDocument8 pagesStandard Operating Procedure For Key Peformance Indicatorsboimzii100% (2)
- A Sample of The Completed Essential Principles Conformity Checklist MD CCLDocument12 pagesA Sample of The Completed Essential Principles Conformity Checklist MD CCLAyman Ali100% (2)
- 93-42-EC ER Checklist ExampleDocument16 pages93-42-EC ER Checklist Exampledulichsinhthai100% (2)
- Gary E. Clayton - Economics. Principles and Practices (2000, McGraw-Hill - Glencoe) PDFDocument664 pagesGary E. Clayton - Economics. Principles and Practices (2000, McGraw-Hill - Glencoe) PDFVictor Camargo100% (2)
- CMA 2020 P1-F AnalyticsDocument63 pagesCMA 2020 P1-F AnalyticsSajan Jose100% (10)
- PMCF PlanDocument2 pagesPMCF Plantarun9917485833% (3)
- Template For PSUR - MPVDocument6 pagesTemplate For PSUR - MPV-MPV Thảo - QAQC100% (2)
- CE Technical File - Medlink IIR Face MaskDocument133 pagesCE Technical File - Medlink IIR Face MaskRicha Rohilla100% (2)
- 1.PMS Plan Template Anexo V. Plantilla Plan de Seguimiento Poscomercialización (PMSP)Document18 pages1.PMS Plan Template Anexo V. Plantilla Plan de Seguimiento Poscomercialización (PMSP)delal karaku100% (2)
- Ethylene Oxide Sterilization Validation ProtocolDocument25 pagesEthylene Oxide Sterilization Validation ProtocolUlisses V A Campos100% (5)
- PMCF ReportDocument6 pagesPMCF Reporttarun99174858100% (2)
- D&D 5e - #07 - Encontros FantásticosDocument30 pagesD&D 5e - #07 - Encontros FantásticosAnonimatoNo ratings yet
- REG.8.2-01-05 Periodic - Safety - Update - ReportDocument12 pagesREG.8.2-01-05 Periodic - Safety - Update - ReportMarcBenetPozo100% (4)
- PMS SopDocument8 pagesPMS Sopstevekent40% (5)
- Literature Search ProtocolDocument11 pagesLiterature Search ProtocolEon Regulatory100% (2)
- Usability Engineering FileDocument8 pagesUsability Engineering Fileeko1980100% (1)
- ClinicalEvaluationReport SampleDocument36 pagesClinicalEvaluationReport Sampleibrahim kademoglu100% (5)
- Periodic Safety Update Report (PSUR) For (Device Name) : Company LogoDocument11 pagesPeriodic Safety Update Report (PSUR) For (Device Name) : Company LogoBo Ram KimNo ratings yet
- Biocompatibility Evaluation For Disposable Oxygen Mask: Document No. Dnocce - 17 Drafted by Reviewed by Approved by DateDocument22 pagesBiocompatibility Evaluation For Disposable Oxygen Mask: Document No. Dnocce - 17 Drafted by Reviewed by Approved by DatePJT Safelock100% (2)
- Periodic Safety Update Report: ProductDocument5 pagesPeriodic Safety Update Report: ProductMauro CostaNo ratings yet
- PMS PMCF CER RelationshipDocument1 pagePMS PMCF CER RelationshipMohammed HammedNo ratings yet
- Sample SoP For Vigilance SystemDocument4 pagesSample SoP For Vigilance Systemsogic100% (5)
- QP19-Vigilance Report - CE MarkDocument18 pagesQP19-Vigilance Report - CE Markanusha shankarNo ratings yet
- EU NB-MED - 2.12 - Rec1 - Rev 11 - Post-Marketing Surveillance - PMSDocument9 pagesEU NB-MED - 2.12 - Rec1 - Rev 11 - Post-Marketing Surveillance - PMSAKSNo ratings yet
- Psur Guidance CDocument16 pagesPsur Guidance CGhada JlassiNo ratings yet
- Post Market Surveillance SOPDocument8 pagesPost Market Surveillance SOPgopinathNo ratings yet
- Technical FilesDocument15 pagesTechnical Fileshitham shehataNo ratings yet
- Quality Statement of SpacexDocument3 pagesQuality Statement of SpacexFaisal AyazNo ratings yet
- 7.post Market Clinical Follow Up ReportDocument2 pages7.post Market Clinical Follow Up Reportdelal karaku100% (1)
- Sop Post Market SurveillanceDocument3 pagesSop Post Market SurveillanceBiolytic LifesciencesNo ratings yet
- EU US PostMarket Surveillance Whitepaper PDFDocument7 pagesEU US PostMarket Surveillance Whitepaper PDFLakshmana Perumal RamarajNo ratings yet
- Post Marketing Surveillance Plan NMRA NewDocument9 pagesPost Marketing Surveillance Plan NMRA NewDeshal RanasingheNo ratings yet
- Post MarketSurveillancePlanTemplateDocument3 pagesPost MarketSurveillancePlanTemplateVomanh HealthcareandFitness100% (1)
- Sop PsurDocument8 pagesSop PsurGehan El Hefney100% (1)
- EU MDR Post-Market Surveillance: Best Practices For Medical Device Regulatory, Compliance & Quality SpecialistsDocument50 pagesEU MDR Post-Market Surveillance: Best Practices For Medical Device Regulatory, Compliance & Quality SpecialistsMauro Costa100% (1)
- Sop VigilanceDocument7 pagesSop VigilanceJane BrownNo ratings yet
- 03 REVAMIL Clinical Evaluation ReportDocument40 pages03 REVAMIL Clinical Evaluation Reportamit545100% (1)
- Post-Market Surveillance - VigilanceDocument40 pagesPost-Market Surveillance - VigilanceSergio Mosa100% (1)
- Medical Devices Benefit Risk ManagementDocument7 pagesMedical Devices Benefit Risk ManagementSteven KingNo ratings yet
- 4.2 Clinical Evaluation Report (RevDocument58 pages4.2 Clinical Evaluation Report (RevMarina Sova100% (2)
- KMDICAClinical Evaluation Report 작성사례집Document216 pagesKMDICAClinical Evaluation Report 작성사례집Suna KimNo ratings yet
- Risk Benefit FinalDocument7 pagesRisk Benefit FinalgoaltechNo ratings yet
- SampleDocument3 pagesSampleccmslaveNo ratings yet
- Psur Apr 2021Document14 pagesPsur Apr 2021Mauro CostaNo ratings yet
- Post Market Surveillance: Global Guidance For Adverse Event Reporting For Medical DevicesDocument37 pagesPost Market Surveillance: Global Guidance For Adverse Event Reporting For Medical DevicesSachin KumarNo ratings yet
- Maintaining Your QMS Under MDR & IVDR - RQSDocument23 pagesMaintaining Your QMS Under MDR & IVDR - RQSliesbeth alberts100% (1)
- Guide Psur Apr 2021Document55 pagesGuide Psur Apr 2021ifrahNo ratings yet
- Medical Device Technical Documentation 1671293832 PDFDocument6 pagesMedical Device Technical Documentation 1671293832 PDFJugurtha Boutlikhet0% (1)
- Spentys - Technical File (Face Shield Mask)Document20 pagesSpentys - Technical File (Face Shield Mask)hitham shehataNo ratings yet
- MEDDEV 2.7.1 Clinical Evaluation Rev 3Document46 pagesMEDDEV 2.7.1 Clinical Evaluation Rev 3Kevin Shane50% (2)
- Risk Assessment 2Document5 pagesRisk Assessment 2Aditya Agrawal100% (2)
- Structure of Technical Documentation PDFDocument4 pagesStructure of Technical Documentation PDFalexgoot100% (1)
- Clinical Evaluation Report SampleDocument12 pagesClinical Evaluation Report Sampleibrahim kademogluNo ratings yet
- 10053865Q00 - PB560 Ventilator & Power Pack Essential Requirements Matrix PDFDocument56 pages10053865Q00 - PB560 Ventilator & Power Pack Essential Requirements Matrix PDFNguyễn Lucy0% (1)
- Clinical Evluation GiudenceDocument20 pagesClinical Evluation GiudenceabcNo ratings yet
- PSUR-PMSR UnterschiedDocument16 pagesPSUR-PMSR UnterschiedwNo ratings yet
- Device Master RecordDocument1 pageDevice Master RecordkuttyjNo ratings yet
- Risk AssessmentDocument26 pagesRisk AssessmentVenkatesh VenkateshNo ratings yet
- PharmacyandPoisonsAct Cap.244Document114 pagesPharmacyandPoisonsAct Cap.244Venkatesh VenkateshNo ratings yet
- ASTM-F3127 Validating Cleaning ProcessDocument18 pagesASTM-F3127 Validating Cleaning ProcessVenkatesh VenkateshNo ratings yet
- EO Annual Requalification ReviewDocument30 pagesEO Annual Requalification ReviewVenkatesh VenkateshNo ratings yet
- ASTMD1238 : Print Collection Join The SeriesDocument14 pagesASTMD1238 : Print Collection Join The SeriesVenkatesh VenkateshNo ratings yet
- Guidance On Change Notification For Registered MD (2dec) For ConsultDocument59 pagesGuidance On Change Notification For Registered MD (2dec) For ConsultVenkatesh VenkateshNo ratings yet
- Composites World 2023 - 07Document52 pagesComposites World 2023 - 07GijoNo ratings yet
- Nfpa 12 2018 14Document1 pageNfpa 12 2018 14Sundar RzNo ratings yet
- Bos 24780 CP 5Document114 pagesBos 24780 CP 5NmNo ratings yet
- BTW IndiaDocument5 pagesBTW IndiaZubair Alam100% (1)
- UntitledDocument22 pagesUntitledGovind UNo ratings yet
- Spa Cip Plus Partial 123Document12 pagesSpa Cip Plus Partial 123Yvonne IngabireNo ratings yet
- Lnvoice Cum Ment Receipt of pAN On 4ea) : AppllcatlDocument4 pagesLnvoice Cum Ment Receipt of pAN On 4ea) : AppllcatlRR Online SevaNo ratings yet
- Delivery Truck Trip DataDocument1,890 pagesDelivery Truck Trip Datatienle.31211024021No ratings yet
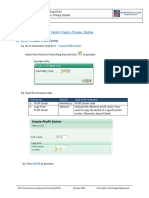
- KE51, KE52, KE53 - Profit Center Create, Change, DisplayDocument5 pagesKE51, KE52, KE53 - Profit Center Create, Change, DisplayElboy Son DecanoNo ratings yet
- Assignment 1 AnswerDocument9 pagesAssignment 1 AnswerpreetNo ratings yet
- Book 2023Document221 pagesBook 2023The WoExPoNo ratings yet
- EEBS2614 FCT1 Memo (RC)Document4 pagesEEBS2614 FCT1 Memo (RC)mohapisthaba77No ratings yet
- Partnership Liquidatio1 LumpsumDocument3 pagesPartnership Liquidatio1 LumpsumJBNo ratings yet
- Importance of Technology in CommunicationDocument4 pagesImportance of Technology in CommunicationHoonchyi KohNo ratings yet
- Setting Up A Variable Capital Company (VCC) in Singapore - Piloto AsiaDocument14 pagesSetting Up A Variable Capital Company (VCC) in Singapore - Piloto Asiaashish poddarNo ratings yet
- London Stock ExchangeDocument9 pagesLondon Stock ExchangeVika IgnatenkoNo ratings yet
- Interloop Annual Report 2023Document190 pagesInterloop Annual Report 2023khizar.alnasir14No ratings yet
- A Survey On Educational Data Mining in Field of Education: Dr. P. Nithya, B. Umamaheswari, A. UmadeviDocument10 pagesA Survey On Educational Data Mining in Field of Education: Dr. P. Nithya, B. Umamaheswari, A. UmadevinpraveenaNo ratings yet
- Corpus Journal of Dairy and Veterinary Science (CJDVS)Document2 pagesCorpus Journal of Dairy and Veterinary Science (CJDVS)Lady CabreraNo ratings yet
- Assignment On Agile MethodologyDocument8 pagesAssignment On Agile Methodologyarisha khanNo ratings yet
- Cta 2&Ft Applied Financial Accounting Reporting 2022 Test 1 - Scenario - TDDocument9 pagesCta 2&Ft Applied Financial Accounting Reporting 2022 Test 1 - Scenario - TDTakudzwa MashiriNo ratings yet
- System Design Implementation and Operation - Chapter 22Document14 pagesSystem Design Implementation and Operation - Chapter 22Chantal DeLarenta100% (1)
- Bahile RCC Citronella Business Proposal - RevisedDocument14 pagesBahile RCC Citronella Business Proposal - RevisedShawn Cassandrabelle Amaris De CastroNo ratings yet
- Department of Management Studies: Geethanjali College of Engineering & TechnologyDocument64 pagesDepartment of Management Studies: Geethanjali College of Engineering & Technologyaurorashiva1No ratings yet
- Market Power: Monopoly and Monopsony: ExercisesDocument12 pagesMarket Power: Monopoly and Monopsony: ExercisesAnnie NguyenNo ratings yet
- Ebook Decision Making in Risk Management Quantifying Intangible Risk Factors in Projects Manufacturing and Production Engineering 1St Edition Cox Christopher O Online PDF All ChapterDocument70 pagesEbook Decision Making in Risk Management Quantifying Intangible Risk Factors in Projects Manufacturing and Production Engineering 1St Edition Cox Christopher O Online PDF All Chapterbeatrice.price716100% (9)