Download as pdf or txt
You might also like
- Geocells in Road ConstructionDocument28 pagesGeocells in Road ConstructionSrinivas J86% (7)
- Crocheted Alien Tutorial JudyDocument4 pagesCrocheted Alien Tutorial JudyJudith Schossböck100% (1)
- Pipeline FrictionDocument17 pagesPipeline FrictionAbelardo ContrerasNo ratings yet
- Intro To Statistical LearningDocument46 pagesIntro To Statistical LearningalvinNo ratings yet
- 5 Regression PDFDocument115 pages5 Regression PDFhawk91No ratings yet
- Math 141: Lecture 18: Correlation and RegressionDocument26 pagesMath 141: Lecture 18: Correlation and RegressionCory DimagibaNo ratings yet
- 330 Lecture15 2014Document53 pages330 Lecture15 2014PiNo ratings yet
- 1 Microbalance Accuracy: Manuel F. G. Weinkauf, José G. Kunze, Joanna J. Waniek, Michal Ku CeraDocument9 pages1 Microbalance Accuracy: Manuel F. G. Weinkauf, José G. Kunze, Joanna J. Waniek, Michal Ku CerawessilissaNo ratings yet
- Regression Analysis - From Statistics To Machine Learning: Ronald Hochreiter Sensational - AiDocument50 pagesRegression Analysis - From Statistics To Machine Learning: Ronald Hochreiter Sensational - AithcNo ratings yet
- Ggplot2 Course2 ch5 SlidesDocument23 pagesGgplot2 Course2 ch5 SlidesSxk 333No ratings yet
- ClusteringDocument62 pagesClusteringRichard RichieNo ratings yet
- Lec1 ppt2019Document23 pagesLec1 ppt2019lcaccompanyNo ratings yet
- 330 Lecture8 2015Document33 pages330 Lecture8 2015Anonymous gUySMcpSqNo ratings yet
- Analyzing The Great Firewall of China Over Space ADocument16 pagesAnalyzing The Great Firewall of China Over Space AManuelNo ratings yet
- Ejemplo de UsoDocument13 pagesEjemplo de UsoCristian Daniel Quiroz MorenoNo ratings yet
- MissingdataDocument10 pagesMissingdataAyaan ShahNo ratings yet
- 330 Lecture8 2014Document34 pages330 Lecture8 2014Anonymous gUySMcpSqNo ratings yet
- LocalGLMnet: A Deep Learning Architecture For ActuariesDocument35 pagesLocalGLMnet: A Deep Learning Architecture For Actuariespapatest123No ratings yet
- Pnas 1811269115 SappDocument25 pagesPnas 1811269115 Sappbahix27973No ratings yet
- Time SeriesDocument19 pagesTime SeriesKIRAN KHANNo ratings yet
- Products For PromotionDocument3 pagesProducts For PromotionVania Diego Velasco RiveraNo ratings yet
- STAT 432: Basics of Statistical Learning: Tree and Random ForestsDocument54 pagesSTAT 432: Basics of Statistical Learning: Tree and Random ForestsRichard AdhyaputraNo ratings yet
- Applied Econometrics WithDocument50 pagesApplied Econometrics WithSamia NasreenNo ratings yet
- Model Visualisation: (With Ggplot2)Document25 pagesModel Visualisation: (With Ggplot2)api-14814295No ratings yet
- Chap1 PDFDocument32 pagesChap1 PDFKevin SánchezNo ratings yet
- 2014 TestDocument13 pages2014 TestAnonymous gUySMcpSqNo ratings yet
- Seppo Pynn Onen Econometrics IDocument22 pagesSeppo Pynn Onen Econometrics Iorxanmeh100% (1)
- Transformations and Misspecification of Econometric Models: August 27, 2014Document19 pagesTransformations and Misspecification of Econometric Models: August 27, 2014Maria RoaNo ratings yet
- Costs: Developing The Language of CostsDocument27 pagesCosts: Developing The Language of Costsee400bps kudNo ratings yet
- 330 Lecture9 2014Document40 pages330 Lecture9 2014Anonymous gUySMcpSqNo ratings yet
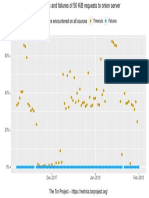
- Problems Encountered On All Sources: Timeouts FailuresDocument1 pageProblems Encountered On All Sources: Timeouts FailuresPatrick PerezNo ratings yet
- Lecture HPC 11 ParallelizationDocument128 pagesLecture HPC 11 ParallelizationAldo Ndun AveiroNo ratings yet
- K-Means RKDocument43 pagesK-Means RKPrince JaiswalNo ratings yet
- Myplot2 PDFDocument1 pageMyplot2 PDFarturdelrioNo ratings yet
- Myplot PDFDocument1 pageMyplot PDFarturdelrioNo ratings yet
- Old Faithful Geyser Data: 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 EruptionsDocument1 pageOld Faithful Geyser Data: 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 EruptionsSantiago ViñanNo ratings yet
- MyplotDocument1 pageMyplotsimuNo ratings yet
- Old Faithful Geyser Data: 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 EruptionsDocument1 pageOld Faithful Geyser Data: 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 EruptionsarturdelrioNo ratings yet
- Old Faithful Geyser Data: 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 EruptionsDocument2 pagesOld Faithful Geyser Data: 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 EruptionsNointNo ratings yet
- Problems Encountered On All Sources: Timeouts FailuresDocument1 pageProblems Encountered On All Sources: Timeouts FailuresPatrick PerezNo ratings yet
- Normal Q Q Plot (Sepal - Length) Normal Q Q Plot (Sepal - Width)Document1 pageNormal Q Q Plot (Sepal - Length) Normal Q Q Plot (Sepal - Width)Carlos MunozNo ratings yet
- ISOMDocument5 pagesISOMAntaraNo ratings yet
- RQ EngellogplotDocument1 pageRQ EngellogplotrojasleopNo ratings yet
- MRT 1550 Vantage Orian Brochure MCAMR0143EADocument19 pagesMRT 1550 Vantage Orian Brochure MCAMR0143EASarpras RumkitNo ratings yet
- Monthly Goal Monthly Log: MON TUE WED THU FRI 1Document1 pageMonthly Goal Monthly Log: MON TUE WED THU FRI 1신희준No ratings yet
- Mutant Year Zero - Zone LogDocument2 pagesMutant Year Zero - Zone LogScott CrumpNo ratings yet
- Zone LogDocument2 pagesZone LogNicolas GalzyNo ratings yet
- Mutant Year Zero - Zone Log PDFDocument2 pagesMutant Year Zero - Zone Log PDFMasis 'Ara' ShahbaziansNo ratings yet
- Zone LogDocument2 pagesZone LogNicolás López CorreaNo ratings yet
- Zone Log PDFDocument2 pagesZone Log PDFКостя СмоляковNo ratings yet
- Zone Log PDFDocument2 pagesZone Log PDFКостя СмоляковNo ratings yet
- Coordinates Terrain Rot Level Threat CommentDocument2 pagesCoordinates Terrain Rot Level Threat Commentfrsayhe5tueayher6uNo ratings yet
- STAT1400 2022 1st Week4-Lecture 7Document19 pagesSTAT1400 2022 1st Week4-Lecture 7Sameer GuptaNo ratings yet
- RQ RegsplineDocument1 pageRQ RegsplinerojasleopNo ratings yet
- Ca FX 4Document1 pageCa FX 4drassuss45No ratings yet
- RQ Mcycle1Document1 pageRQ Mcycle1rojasleopNo ratings yet
- Qqplot 2Document1 pageQqplot 2Sdot BhikangaonNo ratings yet
- Boxplots Degs NSVD - PTL 2Document1 pageBoxplots Degs NSVD - PTL 2Amal KatribNo ratings yet
- Benzecri Distances (2D) RowsDocument1 pageBenzecri Distances (2D) RowsajquinonespNo ratings yet
- Coordinates Terrain Rot Level Threat CommentDocument1 pageCoordinates Terrain Rot Level Threat CommentMasis 'Ara' ShahbaziansNo ratings yet
- Virology, Transmission, and Pathogenesis of Sars-Cov-2: Clinical UpdateDocument6 pagesVirology, Transmission, and Pathogenesis of Sars-Cov-2: Clinical UpdateRocio GmarNo ratings yet
- Joseph D. Challenger, Cher Y. Foo, Yue Wu, Ada W. C. Yan, Mahdi Moradi Marjaneh, Felicity Liew, Ryan S. Thwaites, Lucy C. Okell, Aubrey J. CunningtonDocument17 pagesJoseph D. Challenger, Cher Y. Foo, Yue Wu, Ada W. C. Yan, Mahdi Moradi Marjaneh, Felicity Liew, Ryan S. Thwaites, Lucy C. Okell, Aubrey J. CunningtonRocio GmarNo ratings yet
- Unit 1.1 - Present Simple Present ContinuousDocument5 pagesUnit 1.1 - Present Simple Present ContinuousRocio GmarNo ratings yet
- Mechanisms of Sars-Cov-2 Transmission and Pathogenesis: ReviewDocument16 pagesMechanisms of Sars-Cov-2 Transmission and Pathogenesis: ReviewRocio GmarNo ratings yet
- Present Simple & Present Continuous: I Always Have Breakfast at 7 AmDocument5 pagesPresent Simple & Present Continuous: I Always Have Breakfast at 7 AmRocio GmarNo ratings yet
- CO2 Volume in EggsDocument5 pagesCO2 Volume in EggsFederico LeonNo ratings yet
- AllplanPrecast Product enDocument20 pagesAllplanPrecast Product enCristian MorarNo ratings yet
- Name - Aryan Gupta Reg. No. 199301088 Section - B: Ans.1) A. Rabin Karp String Matching Algorithm CodeDocument7 pagesName - Aryan Gupta Reg. No. 199301088 Section - B: Ans.1) A. Rabin Karp String Matching Algorithm CodebruhNo ratings yet
- Seismic Analysis - Specifying Seismic Weight Through Reference Load CasesDocument4 pagesSeismic Analysis - Specifying Seismic Weight Through Reference Load CasesapirakqNo ratings yet
- Math Sample Paper Class 8Document10 pagesMath Sample Paper Class 8RudraNo ratings yet
- All PastPapersDocument320 pagesAll PastPapersDsquaredNo ratings yet
- Chapter 2-4 To 2-6 With AnswersDocument12 pagesChapter 2-4 To 2-6 With Answersapi-235135985No ratings yet
- Norgren N - UK - 1 - 5 - 135 - PRA - 182000 PDFDocument12 pagesNorgren N - UK - 1 - 5 - 135 - PRA - 182000 PDFpichooooNo ratings yet
- FEMAP-155 508 Cortado (01-13)Document13 pagesFEMAP-155 508 Cortado (01-13)Karolayn VenturaNo ratings yet
- Dempster-Shafer Theory: Introduction, Connections With Rough Sets and Application To ClusteringDocument48 pagesDempster-Shafer Theory: Introduction, Connections With Rough Sets and Application To Clusteringhenri joel jrNo ratings yet
- Maths Decathlon ChallengeDocument41 pagesMaths Decathlon ChallengeSilvens Siga SilvesterNo ratings yet
- bài tập vận dụng chap 1 2 m4bDocument12 pagesbài tập vận dụng chap 1 2 m4bconkienlua2005No ratings yet
- Configuration Manual: AR-SERIES ControllerDocument28 pagesConfiguration Manual: AR-SERIES ControllerJam AnjumNo ratings yet
- Colloids and Surfactants Assignment Title: Micellization: 2 Mphil (Phi Chem.)Document33 pagesColloids and Surfactants Assignment Title: Micellization: 2 Mphil (Phi Chem.)muhammad umairNo ratings yet
- Lifting Lug AnalysisDocument3 pagesLifting Lug Analysisராபர்ட் ஆன்றோ ரெனி67% (3)
- Parameter Estimation of Linear Induction Motor Labvolt 8228-02Document7 pagesParameter Estimation of Linear Induction Motor Labvolt 8228-02Mohamed Riyad BoudallaaNo ratings yet
- Man MachineDocument4 pagesMan MachineAngel Mae PalasNo ratings yet
- Plan and DesignDocument4 pagesPlan and Designsanique peterkinNo ratings yet
- Innovations in Derivatives MarketsDocument446 pagesInnovations in Derivatives Marketsshrikantmsd100% (1)
- R5211001-Electromagnetic Waves and Transmission LinesDocument4 pagesR5211001-Electromagnetic Waves and Transmission LinessivabharathamurthyNo ratings yet
- Professional Competency: Resources Used in Defining Competency GuidelinesDocument19 pagesProfessional Competency: Resources Used in Defining Competency GuidelinesRaul Dolo QuinonesNo ratings yet
- BEF22903 Chapter 2Document35 pagesBEF22903 Chapter 2rajsathia99No ratings yet
- Moment Curvature DiagramDocument3 pagesMoment Curvature DiagramAbera DeressaNo ratings yet
- IEEEpaper Feb 2005Document9 pagesIEEEpaper Feb 2005NicovDijkNo ratings yet
- Prospects of Artificial IntelligenceDocument18 pagesProspects of Artificial IntelligenceSAMEDNo ratings yet
- Proto MathematicsDocument1 pageProto MathematicsthinaNo ratings yet
- Understanding LLDP-MEDDocument3 pagesUnderstanding LLDP-MEDziad Al-showaiterNo ratings yet